Article Text

Abstract
BACKGROUND Submicroscopic subtelomeric chromosome defects have been found in 7.4% of children with moderate to severe mental retardation and in 0.5% of children with mild retardation. Effective clinical preselection is essential because of the technical complexities and cost of screening for subtelomere deletions.
METHODS We studied 29 patients with a known subtelomeric defect and assessed clinical variables concerning birth history, facial dysmorphism, congenital malformations, and family history. Controls were 110 children with mental retardation of unknown aetiology with normal G banded karyotype and no detectable submicroscopic subtelomeric abnormalities.
RESULTS Prenatal onset of growth retardation was found in 37% compared to 9% of the controls (p<0.0005). A higher percentage of positive family history for mental retardation was reported in the study group than the controls (50%v 21%, p=0.002). Miscarriage(s) were observed in only 8% of the mothers of subtelomeric cases compared to 30% of controls (p=0.028) which was, however, not significant after a Bonferroni correction. Common features (>30%) among subtelomeric deletion cases were microcephaly, short stature, hypertelorism, nasal and ear anomalies, hand anomalies, and cryptorchidism. Two or more facial dysmorphic features were observed in 83% of the subtelomere patients. None of these features was significantly different from the controls. Using the results, a five item checklist was developed which allowed exclusion from further testing in 20% of the mentally retarded children (95% CI 13-28%) in our study without missing any subtelomere cases. As our control group was selected for the “chromosomal phenotype”, the specificity of the checklist is likely to be higher in an unselected group of mentally retarded subjects.
CONCLUSIONS Our results suggest that good indicators for subtelomeric defects are prenatal onset of growth retardation and a positive family history for mental retardation. These clinical criteria, in addition to features suggestive of a chromosomal phenotype, resulted in the development of a five item checklist which will improve the diagnostic pick up rate of subtelomeric defects among mentally retarded subjects.
- submicroscopic subtelomeric rearrangements
- clinical preselection
- checklist
- chromosome deletion.
Statistics from Altmetric.com
Mental retardation, the aetiology of which is often unknown, occurs in 2-3% of the general population1 2 and less than 50% of subjects in institutes or schools for children with learning difficulties have a diagnosis.3 4
Chromosome abnormalities are an important cause of mental retardation, detectable in 4-28% of cases, depending on patient selection and laboratory techniques used.4 5 Deletions and translocations larger than 2-3 megabases (Mb) are usually microscopically visible. Some microscopically visible subtelomeric deletions can cause specific malformation and mental retardation syndromes, such as 4p− (Wolf-Hirschhorn), 5p− (cri du chat), 13q−, and 18p− syndromes, and can be ascertained through specific clinical phenotypes. Deletions of other subtelomere regions often have a less characteristic phenotype. In recent years, in situ hybridisation has led to the awareness that submicroscopic subtelomeric chromosome rearrangements are a significant cause of malformation and mental retardation.6 7 Submicroscopic subtelomeric chromosome defects have been found in 7.4% of children with moderate to severe mental retardation and in 0.5% of children with mild retardation of unknown aetiology, both groups of patients with normal G banded chromosomes.7
Some submicroscopic subtelomeric deletions are associated with a specific phenotype. Children with a chromosome 1pter deletion have growth and mental retardation with seizures, visual problems, large anterior fontanelle, asymmetrical and low set, dysplastic ears, deep set eyes, depressed nasal bridge, pointed chin, and fifth finger clinodactyly.8 9 The very rare Ebstein heart anomaly has been observed in two cases with 1p36.3 deletion.8 10
Another example is provided by patients with submicroscopic chromosome 1qter deletions in which, in addition to the growth and mental retardation, severe microcephaly, corpus callosum abnormalities, cardiac abnormalities, hypospadias, and characteristic facies have been described.11 The distal long arm of chromosome 22 is another chromosome which correlates with a phenotype, albeit less obvious.12 The 22q subtelomere deletion is clinically associated with hypotonia, developmental delay, and absence of speech in the child.12 13 This is one of the rare subtelomere deletions that is associated with overgrowth.14
Description of the dysmorphology of individual patients with specific submicroscopic subtelomere deletions is important, as the phenotypes of the different subtelomere deletions are mostly unknown. However, effective clinical preselection is essential because of the technical complexities and cost of screening for subtelomere deletions.
Here, we report a clinical study of 29 mentally retarded children with a known subtelomeric defect and 110 mentally retarded controls with no known diagnosis, but with normal G banded karyotype and without submicroscopic subtelomeric abnormalities.
Material and methods
SUBJECTS AND CONTROLS
Twenty nine mentally retarded persons with a submicroscopic chromosomal rearrangement (18 boys and 11 girls aged 2-20 years, mean age 8.2 (SD 4.8) years) were included. The patients were all referred, for subtelomere testing, to the Institute of Molecular Medicine, Oxford by clinical geneticists from different centres in the UK. Table 1 shows the different subtelomeric abnormalities in the study group. Eleven cases had a de novo abnormality, 17 cases were familial, and one was unknown as the parents were not available for study.
Subtelomeric abnormalities in the study group
A total of 110 mentally retarded persons (69 boys and 41 girls aged 1-19, mean age 8.6 (SD 4.7) years), in whom a subtelomere screen had not detected any abnormalities, were used as controls. All controls had normal routine G banded karyotyping at a 550 band level, and no cause for the developmental delay was found. FMR1gene mutations were excluded where clinically indicated. The controls had been referred from the Institute of Child Health, London (n=68) and from the Department of Clinical Genetics, Oxford (n=42) and had been previously assessed by one of the authors (predominantly RMW for controls from London and JAH for controls from Oxford).
The levels of mental retardation, clinically assessed, in the study and the control groups were: 53% and 53% severe, 36% and 30% moderate, and 11% and 17% mild, respectively. In 19 cases (one subtelomeric and 18 controls) the level of mental retardation was unspecified. All cases were scored by the referring clinical geneticist and the controls by one of the authors (BBAdV), retrospectively from the notes, for clinical variables concerning birth history, growth, facial dysmorphism, congenital malformations, and family history.
FISH ANALYSIS
A total of 5-10 ml peripheral blood was collected from each patient and fixed chromosome suspensions were either prepared directly from the peripheral blood sample or from lymphoblastoid cell lines established from the sample.15 The MultiprobeTM FISH protocol and the subtelomere specific clones used in these studies have been described previously.7 16 17
STATISTICAL ANALYSIS
The data were analysed with version 6.1.3 of SPSS for Windows and the software Confidence Interval Analysis (CIA) written by Gardner and Altman. Comparisons between groups were made using the uncorrected chi-square. Bonferroni corrections were made to assess the significance of the data presented, but actual p values are given in the text.
Results
BIRTH HISTORY AND GROWTH
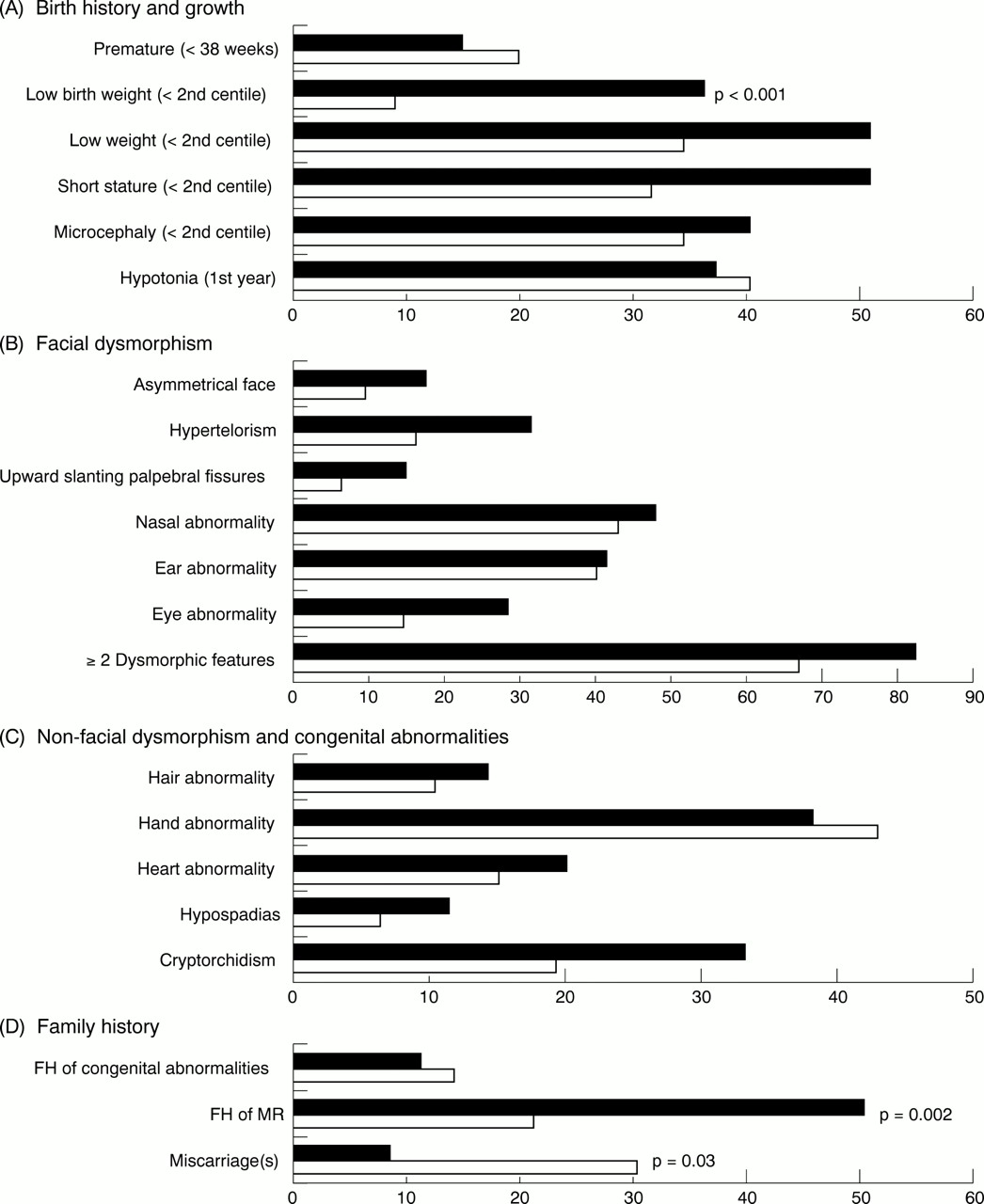
Fig 1A shows the results of birth history and growth in the children with subtelomeric abnormalities compared to the controls. Thirty seven percent (10/27) had a low birth weight (<2nd centile) versus 9% (9/105) of the controls, which is significantly different (p<0.001). This indicates that a child with a low birth weight in our group was more than six times more likely to have a subtelomere deletion (odds ratio 6.3, 95% CI 2.2-17.7). No difference in the degree of birth weight reduction could be observed between the subtelomeric cases and the controls with a low birth weight. Premature births (<38 weeks) are seen in 15% (4/26) of subtelomeric cases. For the other growth variables in the subtelomeric cases, 52% (12/23) had low weight (<2nd centile), 52% (13/25) had short stature (<2nd centile), and 41% (12/29) were microcephalic. Thirty eight percent (11/29) of the subtelomeric cases were hypotonic in their first year of life. None of the latter five features differed significantly from the controls.

The frequencies of features in children with subtelomeric abnormalities (black, n=29) compared to controls (white, n=110) concerning (A) birth history and growth, (B) facial dysmorphism, (C) non-facial dysmorphism and congenital abnormalities, and (D) family history. Only the features with frequencies above 10% are shown.
FACIAL DYSMORPHISM
Fig 1B shows the frequencies of the facial dysmorphic features. Features with a frequency of less than 10% in the subtelomeric cases are not shown. The latter features were ptosis (3%, 1/29), hypotelorism (0%, 0/29), downward slanting palpebral fissures (3%, 1/29), and cleft lip/palate (7%, 2/29).
Several features were common in the subtelomeric cases, including asymmetrical face (17%, 5/29), hypertelorism (31%, 9/29), upward slanting palpebral fissures (14%, 4/29), nasal anomalies (48%, 14/29; notably small/beaked/broad nose, prominent nasal bridge, hyperplastic nasal alae, anteverted nares, prominent columella, prominent philtrum), ear anomalies (41%, 12/29; notably low set/posteriorly rotated/small/prominent/asymmetrical ears, overfolded helices), and eye anomalies (28%, 8/29; notably cataract, strabismus, anophthalmia, microphthalmia, stellate iris, poor vision, posterior embryotoxon). No significant differences in the frequencies of these facial dysmorphic features were seen between the two groups.
Eighty three percent (24/29) of the subtelomeric cases had two or more facial dysmorphic features compared to 67% (74/110) of the controls, which was not significantly different (p=0.10).
NON-FACIAL DYSMORPHISM AND CONGENITAL ABNORMALITIES
Fig 1C shows the frequencies of the non-facial dysmorphic features and congenital abnormalities. Features with a frequency of less than 10% in the subtelomeric cases are not shown. The latter features were skin abnormalities (3%, 1/29), short neck (3%, 1/29), and ambiguous genitalia (0, 0/29).
Renal and brain abnormalities were evaluated adequately in only four and 16 subtelomeric cases, respectively, and five and 62 of the controls, respectively, which made these cases liable to selection. These abnormalities were therefore excluded from further analysis. The following features were observed in more than 10% of the subtelomeric cases: hair abnormalities (14%, 4/29), hand anomalies (38%, 11/29; notably small hands, proximally placed thumbs, trigger thumbs, tapering fingers, clinodactyly, short stubby fingers), heart abnormalities (21%, 6/29; notably PDA, ASD, VSD, subaortic stenosis, bicuspid aortic valve), hypospadias (11%, 2/18), and cryptorchidism (33%, 6/18). No significant differences in the frequencies of these non-facial dysmorphic features and congenital abnormalities were seen between the two groups.
Fifty nine percent (17/29) of the subtelomeric cases had two or more non-facial dysmorphic features or congenital abnormalities or both compared to 47% (52/110) of the controls, which was not significantly different (p=0.28).
FAMILY HISTORY
Fig 1D shows the frequencies of the three variables relating to family history ascertained in the subtelomeric cases and controls. A positive family history for mental retardation was observed in 50% of the subtelomeric cases (14/28) compared to 21% of the controls (23/109), which was significantly different (p=0.002). This indicates that a child with a positive family history for mental retardation in our group was more than three times more likely to have a subtelomere deletion (odds ratio 3.7, 95% CI 1.6-8.9). A positive family history for mental retardation among the subtelomeric cases was found in 14 of the 16 cases with a familial submicroscopic translocation.
A positive family history for congenital malformations was observed in 11% of the subtelomeric cases (3/28) compared to 14% of the controls (15/109) (p=0.67).
Miscarriages were observed in 8% of the subtelomeric cases (2/24) whereas in the controls 30% had a positive family history for miscarriages (33/110) (p=0.03). The latter difference was not significant after the Bonferroni correction. A positive family history for miscarriages among the subtelomeric cases was found in one case with a familial submicroscopic translocation and one case with a de novo abnormality.
The data analysed after stratification for the level of mental retardation did not show other significant differences between the subtelomeric cases and the controls from those presented above (data not shown).
CHECKLIST
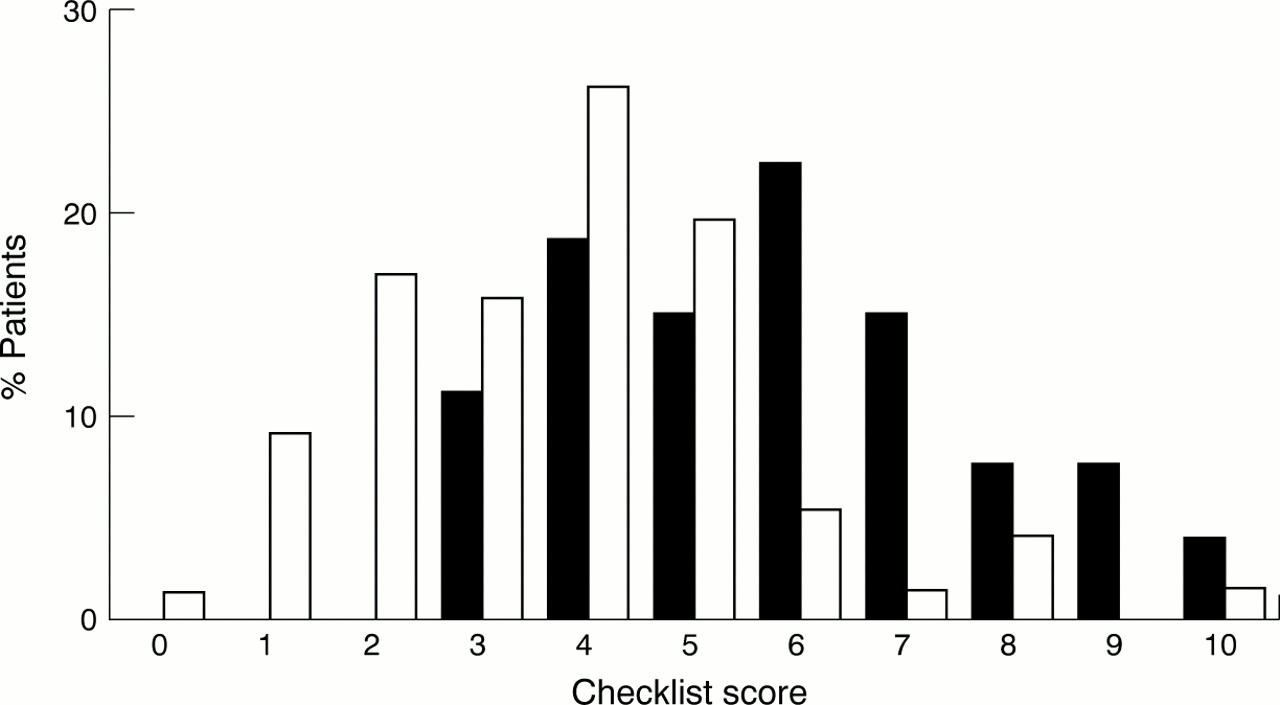
Based on the common features observed in the subtelomeric cases a checklist was developed to help preselection of cases for subtelomere testing. Table 2 shows the five item checklist: (1) family history of mental retardation, (2) prenatal onset growth retardation, (3) postnatal growth abnormalities (either poor or overgrowth), (4) ⩾2 facial dysmorphic features, (5) one or more non-facial dysmorphic feature and/or congenital abnormality. Fig 2 shows the total scores for the five items in 27 of the subtelomeric cases compared to 77 controls. In 35 patients (two subtelomeric cases), one or more of the features (mainly birth weight) were not available and these cases were excluded from the figure and the further analysis for which the total score was required. Using a score of ⩾3 as a cut off for subtelomere testing, one could have excluded 21 of 104 children (specificity 0.27, 95% CI 0.18-0.39) from further cytogenetic testing without missing a subtelomeric case (sensitivity 1.00, 95% CI 0.87-1.00). Table 3 shows the sensitivity and specificity for the different cut off scores. With a cut off score of ⩾4, 36 of 104 children could have been excluded, reducing the number of patients to be tested to 65%, but three of the 27 subtelomeric cases (11%) would have been missed. For a cut off score of ⩾6, the number of tested patients would be reduced to less than a quarter (23%), but almost half of the subtelomeric cases (44%) would have been missed.
Checklist for patients with submicroscopic subtelomeric rearrangements
{kind=link}
{kind=link}
Score for patients with submicroscopic subtelomeric abnormalities (black, n=27) versus controls (white, n=77) using the checklist.
Sensitivity and specificity of the checklist for different cut off scores
Discussion
Since the identification of submicroscopic subtelomeric rearrangements as a major cause of mental retardation,6testing for subtelomeric abnormalities among patients with mental retardation has become an important diagnostic tool. Although widespread screening among mentally retarded subjects might be desirable, the current cost of testing is considered too expensive to allow unselected testing. Moreover, the testing facilities are not at present available to every genetic centre. Therefore clinical preselection is important. So far, selection for subtelomere testing has usually been based on a “chromosomal phenotype” in the patient and/or a family history for the disorder incompatible with monogenetic inheritance.
In the current study, individual clinical features were examined in an attempt to improve preselection of cases. Common features among subtelomeric patients (>30%) in addition to mental retardation were growth retardation (height, weight, and head circumference), hypertelorism, nasal and ear anomalies, hand anomalies, and undescended testes. Although these features are helpful in selecting cases for a subtelomere screen, their frequencies were not significantly different from the controls. This may be because the cases of the control group have been preselected for those features which are a part of the “chromosomal phenotype”. It is inevitable that the clinical geneticists who referred cases for the subtelomere screen (mainly RMW and JAH) did so when they clinically suspected a chromosomal abnormality and normal G banded chromosomes were found. One of those features used in the preselection was microcephaly, which is observed in 37% of the whole patient group. Other studies in large unselected populations of mentally retarded patients in South Carolina and The Netherlands show lower frequencies of microcephaly, 26% and 12% respectively.4 18 In the Dutch study, 31% of the mentally retarded patients had ⩾3 dysmorphic features compared to 48% of the patients in the current study.4 This indicates that in the current study selection of controls has occurred. In spite of this selection, two features did differ significantly between the subtelomeric cases and the controls and should, therefore, be considered as good indicators for subtelomeric abnormalities. These are prenatal onset growth retardation and a positive family history for mental retardation. The low birth weight observed in 37% of the subtelomeric cases had an odds ratio of 6.3 and a positive family history for mental retardation was present in 50% of the subtelomeric cases with an odds ratio of 3.7. The use of both criteria would have increased the pick up rate in our study. However, it should be stressed that 35% of subtelomeric cases (those without a low birth weight or a positive family history) would have been missed if those two indicators had been solely used for selection. A similar situation can be observed if two or more facial anomalies are used for preselection. Remarkably, 18% of the subtelomeric cases in the current study did not have reported facial abnormalities (⩾2) and would therefore have been missed. For example, two previously reported cousins with mild retardation, of which one was in our study group, were facially normal but had a family history which was incompatible with X linked mental retardation. Subtelomere screen showed a submicroscopic monosomy 8p23 and trisomy 20p13.19 As the frequency of subtelomere defects is higher in children with moderate to severe mental retardation compared to mild mental retardation, one might consider limiting the testing to the first group. However, then the three mildly retarded cases (11%) in our study group would have been missed.
The current study shows that making a checklist of clinical features for preselecting cases for subtelomere testing, with high sensitivity and specificity, is difficult. The suggested five item checklist allows 20% of the mentally retarded children to be excluded from testing on clinical grounds. As the controls were selected for a “chromosomal phenotype”, the reduction achieved in an unselected group of mentally retarded children is likely to be higher. The actual specificity of the checklist will only be discovered in a large prospective study of unselected cases with mental retardation who need to be scored and tested for subtelomeric defects. By choosing a higher cut off score one can achieve a higher specificity and thereby limit the number of patients that need to be tested. However, this also means that a number of cases will be missed. For example, a cut off score of 6 and above will reduce the number of patients to be tested to less than a quarter, but almost half of the subtelomeric cases will be missed. Whether this is acceptable to a diagnostic service is very much dependent on the facilities available in each centre.
It is important to stress that the checklist has been used in a relatively small sample of retrospective subtelomeric cases. The phenotypic expression of a subtelomeric defect is dependent on various factors including the size of the deletion, the location of the deletion (which chromosome, short or long arm), the presence of associated trisomy, and the presence of (abnormal recessive) alleles on the remaining homologue. It is plausible that certain small subtelomere deletions with only minor (or even absent) phenotypic effects do exist, which are less suggestive of a chromosomal abnormality, and therefore could easily be missed using the checklist. We may have to accept the latter at present as the cost of subtelomere testing currently limits the number of patients that can be tested. With new developments in the (near) future, such as microarray, which allow more efficient testing, a less stringent inclusion for testing might be possible. Until then selection for the subtelomere test will be required and the presented checklist will be useful.
Acknowledgments
Bert B A de Vries was supported by a grant from the Ter Meulen Fonds (The Netherlands). We would like to thank Dr Esther Crawley for statistical support and Dr Jane Bodden (Worksop) for referring a patient.