Article Text

Abstract
Germline mutations in PTEN can predispose people to Cowden syndrome (CS) and Bannayan-Ruvalcaba-Riley (BRR) syndrome, rare, autosomal dominantly inherited neoplastic disorders. To determine whether germline mutations inPTEN contribute to genetic predisposition to multiple primary tumours within the general population, we conducted a nested case-control study, among 32 826 members of the prospective Nurses' Health Study cohort; cases were women with more than one primary tumour at different anatomical sites. We screened all nine exons of PTEN and flanking intronic splice sites for all 103 eligible cases using SSCP and sequencing. We observed two novel germline heterozygous missense mutations in exon 5 in five of the cases; three were V119L and two were V158L. Neither mutation was observed in 115 controls free of diagnosed cancer (p=0.02). Both mutants showed partial tumour suppressor activity when compared to wild type PTEN when transfected into a PTEN null breast cancer cell line. The phenotype was cell line specific suggesting that genetic background affects growth suppression activity of the mutants. These data provide evidence that germline mutations in PTEN may be a more frequent predisposing factor for cancers in women than previously suggested.
- population based
- tumour suppressor
- multiple cancers
- germline mutations
Statistics from Altmetric.com
PTEN(MMAC1/TEP1),1-3, a tumour suppressor gene located on chromosome 10q23.3, encodes a 403 amino acid dual specificity phosphatase with homology both to the protein tyrosine phosphatase (PTP) family and to cytoskeletal proteins, tensin and auxilin.1-3 PTENhas been shown to be somatically deleted or mutated in a fraction of breast cancers (4-6%),4-6 prostate cancers (35%),7 8 endometrial cancers (35-50%),9 10 glioblastomas (23-44%),11 12 and sporadic melanomas (43%),13 suggesting that it functions as a tumour suppressor gene. A large proportion of tumour associatedPTEN mutations are found in exon 5, the region encoding the phosphatase domain.14 Mutations inPTEN identified in primary tumours, tumour lines, and in patients with the rare inherited cancer syndromes Cowden syndrome (CS) and Bannayan Ruvalcaba-Riley syndrome (BRR), result in ablation of phosphatase activity, showing that enzymatic activity is important for PTEN's ability to function as a tumour suppressor.15 16
Germline PTEN mutations have been identified as the cause of CS, which is characterised by benign adenomas and malignant neoplasms of the breast, thyroid, endometrium, and skin.17 In addition to benign breast disease, adenocarcinoma of the breast develops in approximately 30-50% of women with CS at a mean age of diagnosis 10 years younger than breast cancer in the general population; the lifetime risk of developing epithelial thyroid cancer is 10%. Endometrial carcinoma is part of the spectrum of CS. Inherited PTEN mutations have also been found in BRR,14 18 a rare autosomal dominant disorder characterised by microcephaly, vascular malformations, and benign neoplasms such as lipomas and intestinal hamartomatous polyps. Unlike CS, BRR patients are affected shortly after birth.17 In CS and BRR, 77% of all mutations are found in exons 5, 7, and 8; 43% of all mutations are concentrated in exon 5, which encodes the phosphatase core motif.14
We set out to determine the frequency of germline mutations inPTEN in a population based series of women diagnosed with primary invasive cancer in more than one organ after enrolment in a cohort study. We performed mutational screening of all nine exons and flanking intronic splice sites by SSCP analysis and DNA sequencing. All novel variants that resulted in changes in the encoded protein were functionally characterised.
Materials and methods
STUDY POPULATION
The Nurses' Health Study is a prospective cohort study of 121 700 female registered nurses from 11 US states followed since 1976. Follow up of disease and exposure history has occurred every two years by mailed questionnaire; we seek permission to obtain the medical records from women who report incident cancers. In 1989-1990, blood samples were collected from 32 826 of the cohort members. All women for whom a blood sample was available and who had two confirmed primary incident diagnoses of invasive cancer (other than non-melanoma skin cancer), between 1976 and 1 June 1996, were included in this study. Women who reported cancers before 1976 were excluded from the study, as medical records were not available for review. Controls were randomly selected, cancer free (other than non-melanoma skin cancer) participants and matched for age (±1 year).
MEDICAL RECORD REVIEW AND ASCERTAINMENT OF FAMILY HISTORY
Review of medical records was performed by a physician before mutation analysis and identifying information was then masked. Only women with primary cancers in at least two different organs were included; doubtful diagnoses and metastatic cancers were excluded. First degree family history of breast, melanoma, and colorectal cancer was requested in 1982 and updated in subsequent questionnaires. All links to identifying codes were destroyed and the samples were analysed anonymously; thus, mutational analysis of family members and analysis of LOH in tumours is not possible. In this large geographically dispersed cohort it is unlikely that the subjects are related, but we cannot excluded this. The study protocol was approved by the Committee on Use of Human Subjects of the Brigham and Women's Hospital, Boston, MA, USA.
DNA PREPARATION AND MUTATION SCREENING
Genomic DNA from each subject was prepared using a Qiagen QIAamp 96 Spin Blood Procedure (Qiagen Inc, Chadsworth, CA) for both cases and controls. We performed PCR-SSCP on all nine exons, including flanking intronic sequence, using primers based on previously described sequences.12 Each 50 μl of PCR mixture contained 100 ng of genomic DNA, 1.5 mmol/l MgCl2, 50 mmol/l KCl, 10 mmol/l Tris-HCl (pH 8.3), 0.001% gelatin, 200 mol/l dNTPs, 30 pmol fluorescent labelled primer, and 1.5 U ofTaq polymerase (Amplitaq, Perkin-Elmer Corp). Amplification was conducted using the following cycling parameters: an initial denaturation step at 95°C for five minutes, 35 cycles at 95°C for one minute, 54-58°C for one minute, 72°C for one minute, and a final extension at 72°C for five minutes. Target sequences were amplified using forward and reverse primers labelled with two different fluorescent dyes (6-Fam and Tet) at their 5′ ends. Exon 5 was amplified into two overlapping products for SSCP analysis.
Fluorescent single stranded conformation polymorphism (SSCP) assay was performed on the ABI Prism 377 sequencer using the Genescan 2.1.1 application. To achieve the highest possible mutation detection rate, we applied a set of stringent criteria. In particular, we kept the size of our amplicons to 200-300 bp, used an MDE gel matrix (FMC, Rockland, ME), kept a constant running temperature of 20°C, and used the Genescan 350 ROX (Perkin-Elmer) internal size standard. Known mutantPTEN DNA served as positive controls1 for the SSCP analysis where available.
PCR samples that showed mobility shifts by SSCP analysis were amplified again using exon specific unlabelled primers and purified using the QIAquick PCR Purification Kit (Qiagen, Inc, Chadsworth, CA). Purified PCR products were sequenced directly using Big Dye Terminator cycle sequencing protocol (Perkin-Elmer), electrophoresed on 5% Long Ranger gels (FMC, Rockland, ME), and analysed on an ABI 377 automated DNA sequencer (Perkin-Elmer). Base calling of the sample files was done using the ABI sequence analysis software version 3.0. Factura v 2.0 and Sequence Navigator v 1.01 (Perkin-Elmer) were used to mark potential heterozygous positions and display them for evaluation. Heterozygotes were called at positions where the secondary peak's height was greater than or equal to 45-50% of the primary peak's height in both forward and reverse sequence reads. Where possible, restriction digests with appropriate enzymes were performed to confirm the sequences.
SEQUENCE ALIGNMENTS AND STRUCTURAL ANALYSIS
We examined patterns of sequence conservation inPTEN orthologues using ClustalW. The structural disposition of the mutant residues was determined from the crystal structure of PTEN (PDB entry1D5R)19 and analysed with information from the 1D5R entry in the HSSP database20 to examine possible functional consequences of these mutations.
SITE DIRECTED MUTAGENESIS
A full length PTENcDNA21 in the pZErOTM -2.1 vector (Invitrogen) was used to generate mutants. Mutants were constructed by standard oligonucleotide directed mutagenesis using Pfu polymerase (Stratagene). The PTEN V119L cDNA (M1) was created by PCR using primers 5′-GACAATCATCTTGCAATTCA CTGTAAAG-3′ and 5′-CAGTGAATTGC TGCAAGATTGTCATCTTC-3′. The V158L cDNA (M2) was created using primers 5′-GGGGAACTAAGGACCAGAGACAAAAAG GG-3′ and 5′-GTCTCTGGTCCTTA GTT CCCATAGAAATC-3′. Both constructs were confirmed by sequencing to rule out possible PCR induced mutations. The mutated PTEN cDNAs were then subcloned asNotI fragments into the expression vector pCEP4 for the colony suppression assay.
CELL LINES
Breast cancer cell lines MDA-MB-468, BT-549, and T47D were obtained from American Type Culture Collection. All media were supplemented with 10% fetal bovine serum, 100 units/ml penicillin G, and 100 g/ml streptomycin sulphate.
COLONY SUPPRESSION ASSAY
The generation of pCEP4-PTEN and pCEP4-PTEN-C124S has been described previously.21 Three μg of pCEP4, pCEP-PTEN, pCEP4-PTEN-C124S, pCEP4-PTEN-V119L, or pCEP4-PTEN-V158L were transfected into cells using Fugene 6 (Boehringer-Mannheim). The constructs were transiently transfected into human breast cancer cell lines MDA-MB-468, BT-549, and T47D.21 The MDA-MB-468 and BT-549 lines contain truncating mutations forPTEN which are functionally inactive, whereas the T47D cell line expresses wild type PTEN.1Hygromycin was added to the culture medium after 24 hours to begin selection. Cells were selected over a period of two to four weeks and stained with crystal violet. All experiments were done in triplicate and means were averaged. A two tailed t test was used to test for significant differences between means.
IMMUNODETECTION OF PTEN PROTEIN
For the generation of cell lysates, cells were washed in cold PBS and lysed by incubation at room temperature in a buffer containing 125 mmol/l Tris-HCl (pH 6.8), 10% 2-mercaptoethanol, 4% SDS, and 20% glycerol. Samples were resolved by SDS-PAGE using precast 8-16% Tris-glycine gels (Novex) and transferred onto polyvinylidene difluoride membranes (Immobolin-P, Millipore) for western blotting. Membranes were blocked with Tris-buffered saline containing 0.05% Tween and 5% skim milk and then blotted with anti-PTEN rabbit polyclonal antibody CS486. Blots were developed with horseradish peroxidase conjugated secondary using the enhanced chemiluminescence system (Amersham).
Results
In the overall cohort, 103 women were diagnosed with histologically confirmed primary invasive cancer in at least two different organs between 1976 and 1996 and had given a blood sample in 1989-1990.
Comparisons of cases and controls with respect to year of birth (for which they were matched), family history, and the average age at diagnosis among cases and combinations of cancer sites among cases are presented in table 1. Cases and controls were similar in terms of age and ethnicity; of the 94 cases with known ethnicity 89 were white, reflecting the ethnic distribution of the cohort, four were Asian, and one was Hispanic. The mean age at first cancer diagnosis was 55.5 years and 60.7 years for the second cancer; the majority of women were diagnosed with breast cancer as either their first or second cancer. The average age at diagnosis was 54.0 years for the first cancer and 58.7 years for the second cancer for the five women withPTEN variants. The most common cancer combinations are described in table 1. The tumour spectrum observed may be biased away from the more rapidly fatal cancers, as to be included in the study women diagnosed between 1976 and 1989 had to be alive in 1989 in order to give a blood sample. In comparison to control women without diagnosed cancer, women with multiple cancers had a slightly higher frequency of family history for all types of cancers, but this difference was not statistically significant.
Descriptive characteristics of cases with invasive cancers at more than one anatomical site and controls
MUTATIONAL ANALYSIS
Genotyping using SSCP and sequencing of all nine exons of thePTEN tumour suppressor gene among the cases showed germline heterozygote mutations only in exon 5; no mutations were found in exons 1-4 or 6-9. Five missense mutations (three G→C at codon 119 and two G→C at codon 158) (fig 1) and one silent variant were found in exon 5 among the 103 cases studied (table 2). For the G→C at codon 158 that creates a DdeI restriction site, RFLP analysis confirmed the sequencing results. To determine whether the mutations might represent common polymorphisms, we screened all 115 controls for exon 5 using SSCP and sequencing, and found no mutations (p=0.02 by Fisher's exact test).

Sequence electrophoretograms of PTEN variants found in women with multiple primary tumours. Direct sequence analysis of DNA amplified from the region of exon 5 which contained mutations. (A, C) Arrows point to the heterozygote, G/C, (shown in the reverse complement) mutation found at codon 158 (GTA/CTA). This mutations creates a DdeI restriction site. (B) DNA sequence of homozygous wild type.
Summary of PTEN variants in women with multiple primary tumours
Three women (samples 3, 4, 5) had a Val→Leu substitution at codon 119 (M1) (within the phosphatase domain) and two women (samples 1 and 2) had a Val→Leu substitution at codon 158 (M2) (C-terminal to the phosphatase domain) (table 2). One woman (sample 2) had an additional silent variant at codon 130. Four of the five women (samples 1, 2, 4, and 5) with an exon 5 mutation had developed breast cancer. Endometrial cancer was diagnosed in two of the three women with mutations in codon 119; one of these women (sample 3) also had ovarian cancer, and another (sample 4) had a history of lung adenocarcinoma and ovarian teratoma. Samples 2 and 5 were the only two women with a family history of cancer in a first degree relative (table 2). The women are self-identified whites; further details of ancestry in the five women with thePTEN variants is unknown.
Cowden syndrome was not mentioned in any of the medical records; in particular, there was no mention of CS associated skin lesions or hamartomas. Although two of the women reported benign colon polyps and one reported benign uterine fibroids, from the limited information available, they did not meet the criteria for diagnosis of Cowden syndrome,17 although we cannot exclude this diagnosis as we were not able to conduct a standardised physical examination.
STRUCTURAL AND PHYLOGENETIC ANALYSIS OF VARIANT RESIDUES
Sequence alignments showed that the valine at position 119 (M1) is conserved in PTEN homologues and other phosphatases down to yeast and the valine at position 158 (M2) is conserved in PTEN homologues among vertebrates. In the PTEN structure, both mutant residues are located in the catalytic domain, about 7 angstroms and 9 angstroms, respectively, from residues involved in the phosphatase catalytic site as defined by a match to the PROSITE database entry PS00383. Furthermore, the residue mutated in M1 is inaccessible to solvent. The residue mutated in M2 is within 5 angstroms of residues 14, 15, and 159. These residues are all conserved among proteins sharing at least 30% sequence identity withPTEN, as reported in the HSSP database entry corresponding to the PTEN structure (1D5R).20 In this same multiple alignment, the variant leucine amino acid is not found at either position 119 or 158. Instead, only valine, isoleucine, and threonine are found at position 119, while lysine, valine, isoleucine, and arginine are found at position 158. Given the buried disposition of residue 119, the proximity of the mutant residues to thePTEN active site, and the exclusion of leucine from either position in the phylogenetic comparisons, the mutations, especially V119L, might be disruptive toPTEN structure or function or both.
TUMOUR SUPPRESSOR ACTIVITY
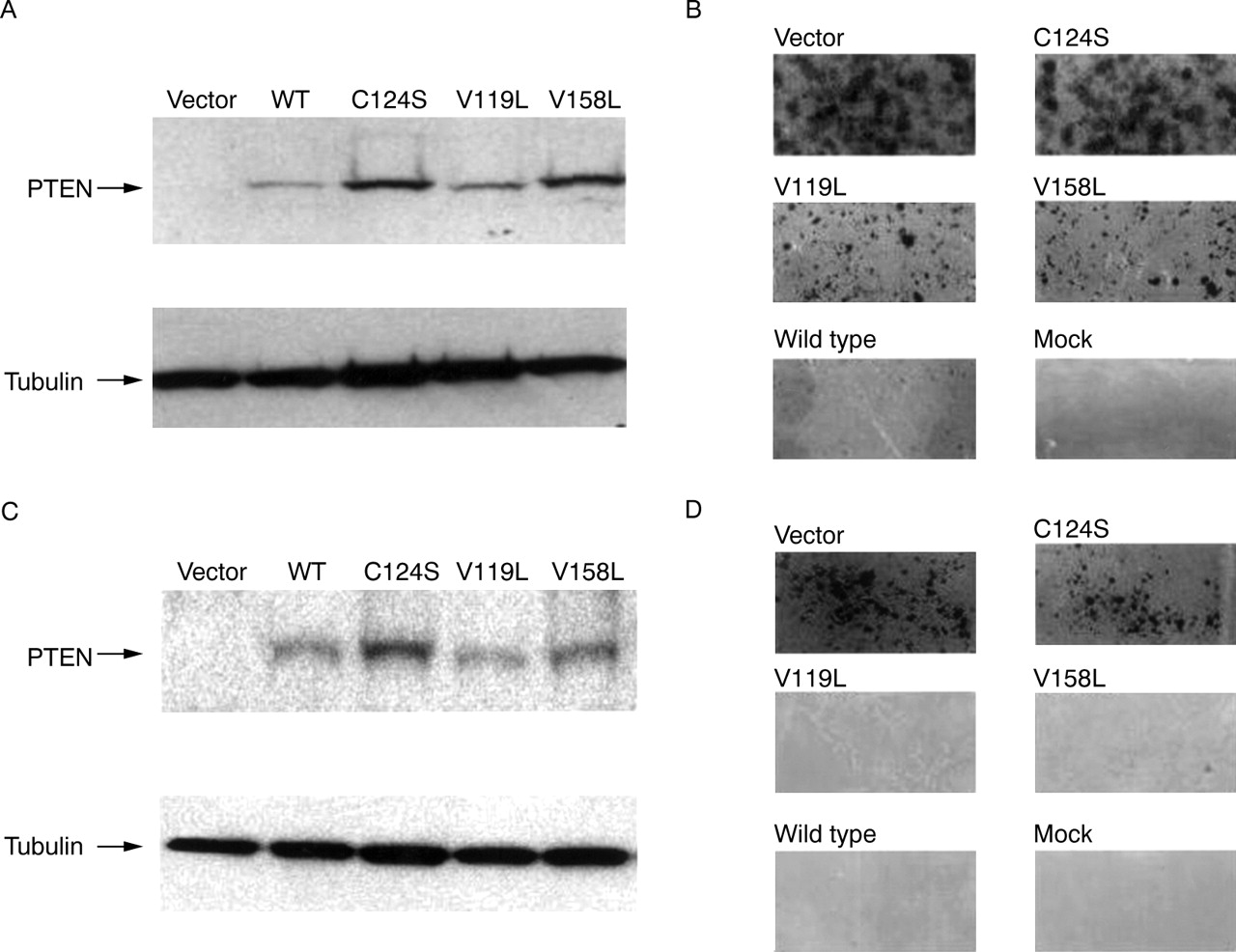
To investigate the possibility that these new variants may impair the tumour suppressor function of PTEN, we examined these mutants in a standard colony suppression assay21 using the selection agent hygromycin and vectors expressing wild type PTEN (pCEP4-PTEN), phosphatase dead PTEN (pCEP4-PTEN-C124S), and the mutants M1 (pCEP4-PTEN-V119L) and M2 (pCEP4-PTEN-V158L). All transfected cell lines expressed comparable levels of wild type and mutant PTEN as detected by immunoblotting (fig2, panels A and C). Examination of colony formation in MDA-MB468 (fig2, panel D) and T47D (data not shown) showed that M1 and M2 mutants formed fewer colonies than vector alone (p<0.01) but were not statistically different from wild type PTEN (table 3). Colonies formed by cells transfected with the empty vector pCEP4 were numerous and large, comparable to those formed by cells expressing the functionally inactive PTEN construct pCEP4-PTEN-C124S. Colony formation was inhibited in those cell lines expressing the wild type PTEN construct, pCEP4-PTEN. The M1 and M2 expressing cells did form colonies in cell line BT-549 (fig 2, panel B), though significantly (p<0.001) fewer in number and not as robust compared to the colonies formed with the vector alone or with the PTEN-C124S mutant, but clearly different (p<0.001) from those expressing wild type PTEN (p<0.001) (table 3).
{kind=link}
{kind=link}
PTEN expression and growth suppression in breast cancer cell lines. (A, C) Detection of PTEN protein after transfection. Western analysis using anti-PTEN or anti-β-tubulin antibodies on total cell lysates from the PTEN null cell lines, BT-549 (A) and MDA-MB-468 (C) after transfection. Arrows indicate the position of PTEN and β-tubulin. Cells were transfected with empty pCEP4 vector (Vector) and pCEP4 vectors containing wild type PTEN (WT), PTEN-C124S (C124S), PTEN-V119L (V119L), and PTEN-V158L (V158L). Approximately equal amounts of lysate are present in all lanes as indicated by the tubulin expression. (B, D) Measurement of colony suppression by PTEN. Cells were transfected with pCEP4 based constructs (vector, wild type, C124S, V119L, V158L) that contain the hygromycin resistance cassette or the vector CMV-β-galactosidase (mock) which lacks the hygromycin resistance cassette and grown with media in the presence of 200 mg/ml of hygromycin. After 14 days no cells were present in the mock flasks. Large colonies grew in flasks transfected with pCEP4 vector and C124S. For BT-549 (B), V119L and V158L colonies were appreciably larger than wild type but smaller than the colonies seen with vector and C124S. For MDA-MB-468 (D), V119L and V158L colonies were similar in size and number to wild type. Colonies from vector and C124S were much more numerous and larger than the colonies of V119L, V158L, and wild type.
Mean colony counts and size for phosphatase inactive PTEN, mutant PTEN, and wild type PTEN constructs transfected into PTEN null breast cancer cell lines
Discussion
We report two novel functional germline mutations within exon 5 of the tumour suppressor gene, PTEN. To our knowledge, this is the first population based study that describes germline PTEN mutations in women with multiple primary tumours at various sites. To date, these specific mutations have not been observed in published Cowden pedigrees.
The mutations we observed occur in evolutionarily conserved positions. Results from the colony suppression assay show that the mutations impair the tumour suppressor activity ofPTEN. The fact that the mutants V119L and V158L give an intermediate phenotype suggests that these mutants may have partial activity or altered function. Previously, PTEN protein with the CS mutation, G129E, has been shown to have normal phosphatase activity against protein substrates both in vitro22 and in cell lines,16 but has no phosphatase activity against Ptd-Ins (3,4,5) P3, suggesting that these two activities have different protein sequence requirements. Evidence from the colony suppression assay suggests that we have identified hypomorphic alleles that alter but do not abolish PTEN's function. Also, colony formation was cell line specific; colonies formed only with BT-549, suggesting that a specific genetic/cellular milieu is required to elicit a phenotype with these mutations. The implication is that germline mutations in PTEN may not always result in a cancer phenotype, but instead may require a specific genetic background for complete penetrance. In contrast to other tumour suppressor genes, such as BRCA1 where the penetrance in breast cancer pedigrees is high (up to 85%) for developing breast cancer,23 women affected with CS have only a 30-50% lifetime risk of developing breast cancer.17 This is similar to more recent estimates of penetrance of mutations in BRCA1 derived population based studies.24 25 First degree family history of cancer was only present in two of the five women withPTEN variants, consistent with a relatively low penetrance of these specific mutations. The fact that the same mutations were found among women with different tumours is not surprising as identical mutations in PTENhave been found in both CS and BRR.26 Modifier genes or stochastic effects may be responsible in part for the different phenotypes occurring in the cases with identical mutations (V119L, samples 3, 4, 5 and V158L samples 1, 2). Recent analyses of different lines of mutant mice suggest that genetic background can significantly affect PTEN phenotypes.27-29 Though the heterozygote mice all showed an increase in tumour incidence and hyperproliferative lesions of the intestines, the spectrum of other hyperproliferative and neoplastic disorders differed dramatically between lines.
Estimates of the frequency of CS range from 1 in 1 000 00017 to 1 in 250 000.30 Among people with a clinical diagnosis of CS, the PTENmutation prevalence ranges from 13 to 80%.17 31 The contribution of germline mutations in PTENtowards genetic predisposition to cancers within the general population is for the most part unknown. A recent study of early onset breast cancer patients reported germline missense mutations in two of 60 patients; one of the two mutations was in exon 5.32 Rheiet al 5 reported one germline mutation in 54 breast cancer patients, and this single patient was thought to have clinical CS. In a series of 64 non-CS cases ascertained with the minimum diagnosis of breast cancer and thyroid pathology, one proband was found to have a germline mutation inPTEN. In addition to breast and thyroid cancers, this person had endometrial cancer.33 We report aPTEN mutation prevalence of 5% in a series of 103 women with multiple primary tumours selected from 32 826 women who gave us a blood sample. PTEN has been considered to be the susceptibility gene for rare autosomal dominantly inherited multiple cancer syndromes. Our results suggest that either CS is substantially underdiagnosed or that germline mutations are a predisposing factor for multiple tumours occurring in the absence of CS.
Acknowledgments
This work was supported by NIH grants CA40356, CA65725, CA70817, and CA49449. We would like to thank Jeanne Sparrow, Lisa Li, Gary Gearin, and Michael L Fitzgerald for their technical assistance, and Barbara Egan for disease follow up documentation. We would like to thank Daniel Chasman for help with analysis of crystal structure. We are also indebted to the participants in the Nurses' Health Study for their dedication and commitment.