Article Text

Abstract
We report on a familial submicroscopic translocation involving chromosomes 8 and 16. The proband of the family had a clinical picture suggestive of a large deletion in the chromosome 16p13.3 area, as he was affected with tuberous sclerosis complex (TSC) and had α thalassaemia trait, and his half brother, who also had TSC, may have suffered additionally from polycystic kidney disease (PKD). FISH studies provided evidence for a familial translocation t(8;16)(q24.3;p13.3) with an unbalanced form in the proband and a balanced form in the father and in a paternal aunt.
The unbalanced translocation caused the index patient to be deleted for the chromosome 16p13.3-pter region, with the most proximal breakpoint described to date for terminal 16p deletions. In addition, FISH analysis showed a duplication for the distal 8q region. Since the index patient also had hypomelanosis of Ito (HI), either of the chromosomal areas involved in the translocation may be a candidate region for an HI determining gene. Furthermore, it is noteworthy that both carriers of the balanced translocation showed a nodular goitre, while the proband has hypothyroidism.
- PKD1
- TSC2
- HI
- partial trisomy/monosomy
Statistics from Altmetric.com
We report on a patient affected with tuberous sclerosis complex (TSC), α thalassaemia trait, and hypomelanosis of Ito (HI), whose half brother, also affected with TSC, had died from renal complications.
Tuberous sclerosis complex (TSC) is an autosomal dominant disease characterised by skin lesions, mental handicap, seizures, and the development of hamartomas in many different organs and tissues. TSC determining loci have been mapped to chromosome 9q34 (TSC1) and 16p13.3 (TSC2). Both have been identified and characterised.1 2 The major gene for autosomal dominant polycystic kidney disease (PKD1) has also been identified and maps to chromosome 16p13.3, proximally adjacent to the TSC2 locus.3 A number of patients with large deletions disrupting both theTSC2 and the PKD1genes who have TSC and childhood onset polycystic kidney disease have been reported.4 The α globin gene cluster has also been mapped to chromosome band 16p13.3 distal to theTSC2 locus. Deletions, variable in extent but limited to the terminal part of band 16p13.3, were reported in patients with α thalassaemia/mental retardation syndrome (ATR-16).5 Apart from the haematological abnormality presenting as either Hb H disease or α thalassaemia trait, the other clinical findings of this condition are mild to moderate mental handicap and a broad spectrum of associated dysmorphic features. Therefore, TSC2,PKD1, and ATR-16could be involved in a contiguous gene syndrome with characteristic phenotypic abnormalities. This was suspected in the patient reported here.
Hypomelanosis of Ito (HI) is a neurocutaneous disorder characterised by hypopigmented whorls, streaks, and patches typically distributed along the lines of Blaschko; one or more abnormalities of the central nervous system, eyes, hair, teeth, and musculoskeletal system have also been described in about 70% of HI patients. The genetic basis of HI is still debated but published data indicate the existence of a wide variety of chromosomal mosaicism in different cases. In addition, a subgroup of affected females with non-mosaic balanced X;autosome translocations have been reported.6 The phenotype in these cases appears to be related to the presence of mosaic functional disomy rather than a disruption of an X linked gene. From these data it appears that there is no common genetic background to this syndrome.
In our patient the presence of clinical findings clearly indicative of HI, in combination with findings suggestive of a contiguous gene syndrome involving the distal chromosome 16 region, has prompted us to evaluate the genetic background of this association. In this paper, the clinical findings and the cytogenetic and molecular analyses of the patient and his relatives are reported.
Case report
The male proband (fig 1) was born at term after a normal pregnancy and delivery. His birth weight was 2500 g. The parents were healthy and unrelated. The proband's paternal half brother was diagnosed as having TSC (fig 2). Besides cortical tubers and subependymal nodules, brain MRI showed a Dandy-Walker malformation. He died at 18 years of age following intraperitoneal haemorrhage owing to renal angiomyolipoma.

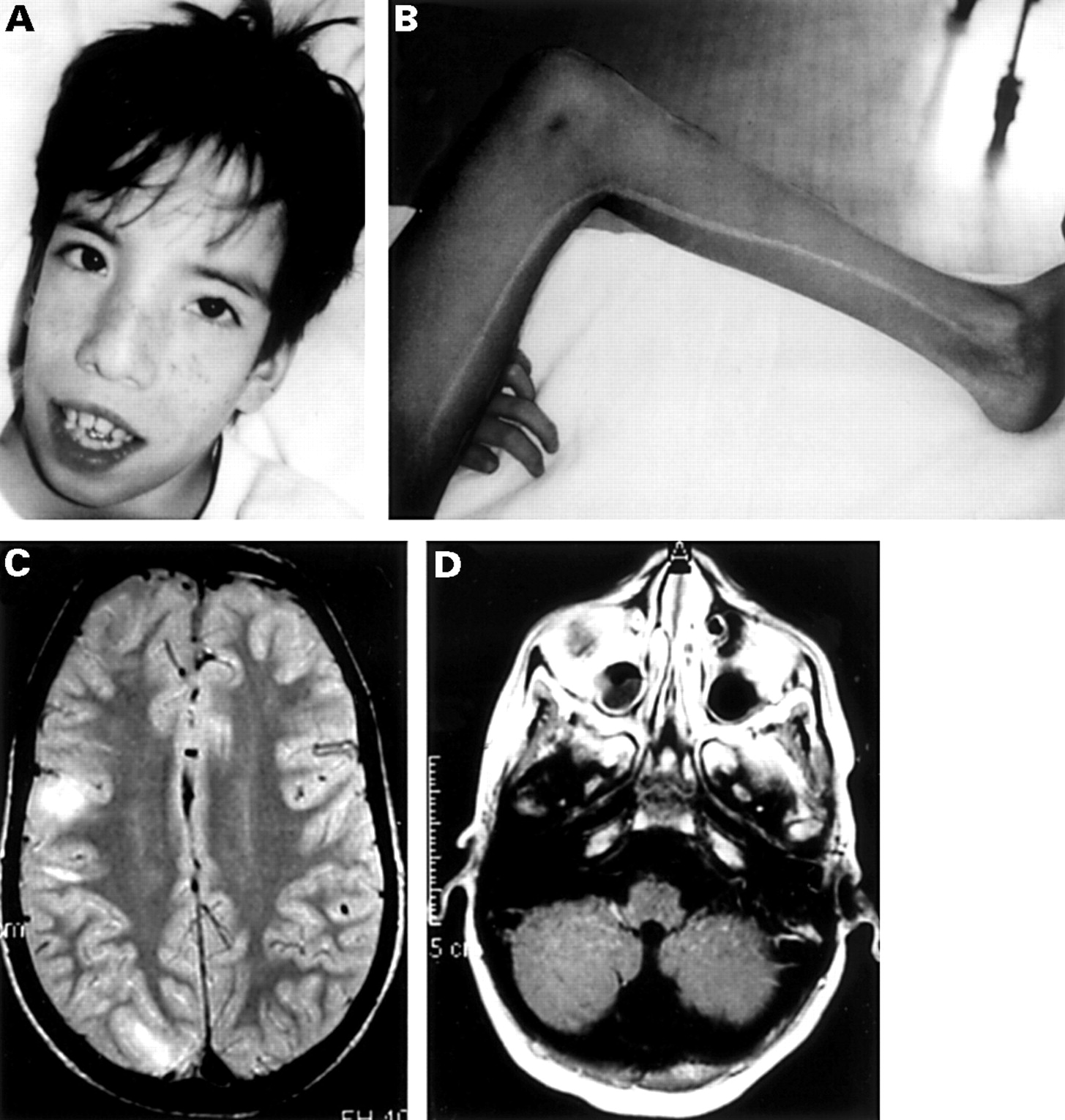
The proband (III.3) showing (A) facial angiofibroma and dysmorphic facial features and (B) a hypopigmented streak on his left inner leg. (C) The brain MRI scan showed multiple cortical and subcortical tubers and (D) the presence of a Dandy-Walker malformation.
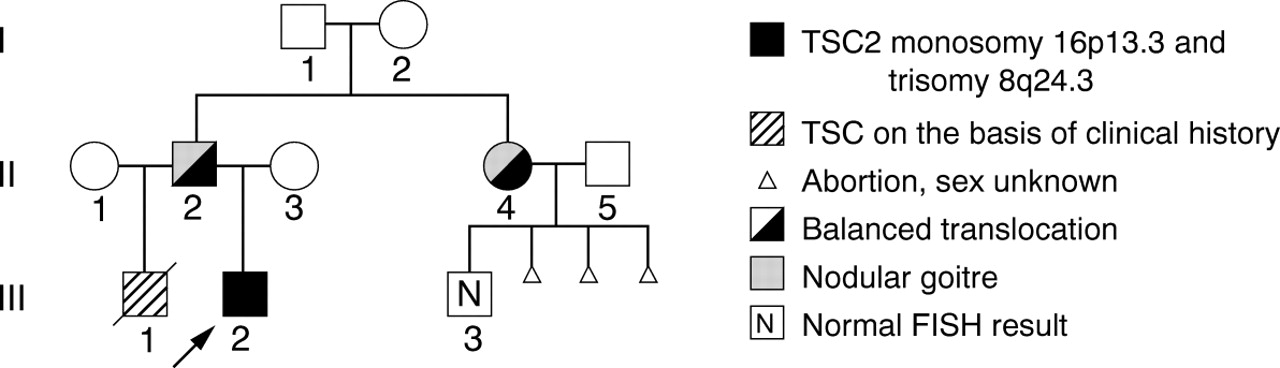
Pedigree of the family with t(8;16)(q24.3;p13.3).
At birth, the proband was noted to have omphalocele, dysmorphic face, and left talipes equinovarus, requiring corrective plaster casts until the age of 6 months. At the age of 3 years, the diagnosis of TSC was made on the basis of the presence of hypopigmented skin spots, mental retardation, cortical tubers, and subependymal nodules on the brain CT scan. From 11 years of age, he developed complex partial seizures which were successfully treated with carbamazepine. At this time, physical examination showed facial angiofibromas, fibrous forehead plaque, three hypopigmented spots, dysmorphic facial features with a triangular face, high forehead, hypertelorism, epicanthic folds, broad nasal root, narrow and high arched palate, hypertrophic alveolar ridges, supernumerary teeth, microcephaly, and a generalised muscular hypotrophy (fig 1A). The left leg was 2 cm shorter than the right.
He had a well demarcated 1.5 cm wide hypopigmented streak striating from the left groin and extending linearly to the medial malleolus (fig1B). Another irregular hypopigmented area was present on the left lower abdomen. Psychomotor development was severely delayed. Haematological investigations showed mild hypochromic microcytic anaemia (Hb 9.8 g/dl, MCV 65.8 fl, MCH 22.2 pg) with normal levels of Hb A2. Ophthalmological examination and cardiac and abdominal ultrasound were normal.
Brain MRI showed multiple cortical and subcortical tubers and one periventricular subependymal nodule. Generalised cerebral atrophy with enlarged lateral ventricles and inferior vermian hypoplasia were also observed (fig 1D).
Radiographs of the lower limbs confirmed that the right femur was 2 cm longer than the left, with an area of osteosclerosis in the right distal femur.
Follow up renal ultrasound at the age of 12 years 6 months showed bilateral multiple renal cysts, the largest of which was 10 mm in diameter. These findings were confirmed by renal CT scan.
No pigmentary abnormalities were present in the patient's parents. His father showed no signs of TSC after extensive examination, which included fundoscopy, renal and cardiac ultrasound, and brain MRI. He did have symptoms of hyperthyroidism; a nodular goitre was diagnosed. Recently, a nodular goitre with normal thyroid function was also diagnosed in the father's sister (fig 2, II.4), who was otherwise healthy without signs of TSC. In the proband, laboratory investigations showed high TSH and low T4 levels; thyroid echography was normal.
Materials and methods
CYTOGENETICS
Chromosome analysis was performed according to standard procedures with GTG banded chromosomes from cultured peripheral blood lymphocytes. The slides were used directly for FISH.
DNA ANALYSIS
DNA was isolated from peripheral blood samples using the salting out method.7 Southern blot analysis was done onHindIII or EcoRI digested DNA with probes (4B2, E0.7, and 1A1H.6) from theTSC2 cDNA1 labelled by the random primer method. The VNTR at the α globin locus (3′HVR) was analysed after PstI digestion. In addition, the microsatellite markers KG8, 16AC2.5 (D16S291), and SM7 (D16S283)1 3 were analysed.
FISH ANALYSIS
For routine TSC2 deletion screening, the plasmid CW23 (3′ TSC2 gene) and the cosmid CW9D, about 30 kb distal to the TSC2gene, were used1 (fig 3G).

{kind=link}
{kind=link}
{kind=link}
(A) In situ hybridisation on the proband's metaphases (fig 2, III.3) shows a deletion of the 16p13.3 region with CW9D (arrowhead) and (C) a partial trisomy for the 8q24.3 region with probe 2053b (arrowhead). (B) Metaphases of the proband's father show a balanced translocation t(8;16)(q24.3;p13.3) with the probes CW9D (16p13.3) (arrow) and 2053b (8q24.3) (arrowhead). (D) The breakpoint on 16p13.3 is visualised by the probe 1.8F, with signals present on 16p13.3 as well as on 8q24.3 (arrowheads). (F) Probe CHT 16 (5′ TG gene) remains on 8q24.3 (green signals), while the cosmid CW9D is translocated from 16p13.3 to 8q24.3 (red signals). (E) Ideogram of the cryptic translocation t(8;16)(q24.3;p13.3). (G) The molecular map of the translocation area is not drawn to scale. Probe 1.8F crossing the breakpoint is highlighted by a red bar. The vertical arrowhead indicates the breakpoint of the t(16;22)(p13;q11) as analysed by The European Polycystic Kidney Disease Consortium 1994.3
The breakpoint analysis of the 16p13 region was performed with the cosmids from the PKD1 region LA3 (N54)1 and cGGG4b (GGG12).8 In addition, PACs 97.10G, 1.8F, and 96.4b,9 located proximal to thePKD1 gene, were used.
For the 8q24 breakpoint analysis, we used the following probes (fig3G): cMYC,10 Drg1 PAC 28N23,11 TG gene cosmids CHT 3, 11, 16, and 18,12 and the subtelomeric probe 2053b3.13The orientation of the TG gene and theTG cosmids is indicated in fig 3G (F Baas, L Kalaidijeva, unpublished results).
For the identification of chromosome 8, we used the α satellite centromere probe αp8 (D8Z1)15 directly labelled with tetramethyl-rhodamine-dUTP using the nick translation kit (Boehringer Mannheim). All other probes were labelled either with biotin or digoxigenin with the BIO- or DIG-NICK kit (Boehringer Mannheim).
FISH was performed according to standard protocols.15 16 After an overnight hybridisation at 37°C and washing to a stringency of 0.1 × SSC at 55°C, detection was performed by a single detection layer, either anti-DIG-FITC (Boehringer Mannheim) or streptavidin-Alexa-594 (Molecular Probes) in a low salt buffer. Slides were mounted with Vectashield (Vector) containing 4′,6′diamino-2-phenylindole (DAPI) or propidium iodide (PI) (Sigma). FISH signals were captured and analysed with a Powergene workstation (PSI Chester) mounted on a Leica DMRXA microscope.
Results
Cytogenetic analysis of both the index patient and his father showed normal male karyotypes (results not shown). The combination of TSC and the haematological findings in the proband prompted the search for a large, but submicroscopic deletion in the chromosome 16p13.3 region harbouring both the TSC2 gene and the α globin locus. Southern blot analysis with the cDNA probes 4B2, E0.7, and 1A1H.6, covering the entire coding region of theTSC2 gene, suggested a deletion spanning at least the TSC2 gene. This was confirmed by FISH analysis using probes CW23 (3′ part of theTSC2 gene) and CW9D (about 30 kb upstream of the gene) (fig 3A). Analysis of the VNTR at the α globin locus (3′HVR) showed heterozygosity in the patient's father and hemizygosity in the proband with absence of a paternal allele. On the centromeric side, the proband showed a normal heterozygous pattern at D16S291 and SM7, whereas the microsatellite marker KG8 (3′ end of thePKD1 gene) was uninformative. Hence, we found an apparently de novo deletion of paternal origin including theTSC2 gene and extending into or including the α globin locus on the telomeric side and the possibility of involvement of the PKD1 locus on the centromeric side. Since the father had lost a previous child with TSC, the possibility of gonadal or low grade gonosomal mosaicism was considered. To explore the latter possibility, FISH studies were performed. In parallel, a further delineation of the breakpoint area on the centromeric side was attempted.
The probes LA3 (N54) and cGGG4b (GGG12) were shown to be involved in the deletion, thereby implicating the PKD1gene. In the father's metaphases, no cells carrying a deletion could be identified. Instead, normal signals of the chromosome 16 probes were found both on one chromosome 16p13.3 and on one chromosome 8 in the qter region. The translocation was shown to be reciprocal as the subtelomeric chromosome 8 probe 2053b3 was found on the der(16) (fig3B-E). The other chromosome 8 loci tested,MYC, Drg1(D8S557), as well as the thyroglobulin (TG) locus (tested with the CHT 3/11 and CHT 16/18 probes for the 3′ and 5′ ends of the gene), were not involved in the translocation (fig 3F). The demonstration of a balanced translocation in the father allowed a further refinement of the breakpoint area on chromosome 16, as FISH with the PAC clone 1.8F showed signals on both der(8) and der (16) (fig3D).
The unbalanced form of the translocation in the proband was further characterised with 2053b3, which showed signals on the der(16) as well as on both chromosomes 8 (fig 3C). Hence, the resulting abnormality in the proband is a deletion of the chromosome 16 region telomeric to the breakpoint in PAC 1.8F, including the PKD1,TSC2, and α globin genes and a duplication of the chromosome 8q24.3-qter region.
Subsequently, FISH analysis was applied to analyse paternal relatives at risk. The father's sister (II.4) was also shown to be a carrier of the balanced translocation, whereas her son (III.3) showed normal signals with the breakpoint clone 1.8F.
Discussion
Contiguous gene syndromes are an important issue in clinical genetics. Keen clinical observation has contributed to the delineation of syndromes such as Kallman syndrome, DiGeorge syndrome, and Miller-Dieker syndrome.
Several contiguous gene syndromes involving the chromosome 16p13.3 region have been described. The combination of α thalassaemia, dysmorphic features, and mental retardation, ATR-16, is caused by a terminal deletion of chromosome 16p13.3 with the breakpoint distal to the adjacent TSC2 andPKD1 genes.5 A TSC patient with partial monosomy of this region has been described, whose deletion included the TSC2 gene, with the breakpoint disrupting the PKD1 gene (fig 3G), as a result of the unbalanced form of a familial translocation t(16;22)(p13.3;q11).3 Relatives who carried the balanced form of the translocation were affected with adult polycystic kidney disease and the analysis of this family has been instrumental in the identification of the PKD1 gene. Recently, several patients with TSC and early onset polycystic kidney disease were shown to have deletions involving both theTSC2 and PKD1genes.4 The proband of the family described here had a clinical picture suggestive of a large deletion in the chromosome 16p13.3 area, as he was affected with TSC and had α thalassaemia trait. The death of his half brother, who also had TSC, at 18 years of age owing to renal complications indicated the possibility of involvement of the PKD1 gene. FISH studies provided evidence for a familial cryptic translocation t(8;16)(q24.3;p13.3) with an unbalanced form in the proband and a balanced form in the father and paternal aunt. The unbalanced translocation caused the index patient to be monosomic for the chromosome 16p13.3 region with a breakpoint detected by PAC 1.8F, which represents the most proximal breakpoint described to date for terminal 16p deletions. In addition, FISH analyses showed trisomy for the distal 8q region as a result of an adjacent 1 segregation.
To our knowledge, constitutional translocations involving 8q24.3 are very rare,17-19 presumably because they escape detection by routine cytogenetic techniques. A specific phenotype associated with a partial trisomy 8q24-qter has not been defined.18 19 In contrast, balanced and unbalanced rearrangements including 8q24 have been noted in a variety of haematological and solid tumours.20
The thyroid dysfunction, which was diagnosed in both unbalanced and balanced translocation carriers, is possibly of interest, because the chromosome 8q24.3 region harbours the thyroglobulin (TG) gene. However, since in all three patients, both the 3′ and the 5′ end of theTG gene were shown to remain on chromosome 8 by FISH analysis, disruption of the coding region of the gene seems unlikely.
The complex clinical picture of the proband can be explained to some extent by deletion of known or as yet unknown genes in the chromosome 16p13.3 region, as he clearly shows symptoms of both TSC and ATR-16. Since the PKD1 gene is also deleted, it is noteworthy that until now severe renal disease has not developed. Because the index patient has also been diagnosed with HI, this study has contributed to resolving the genetic background of HI in this patient. The patient with the unbalanced t(16;22) translocation, described in the paper reporting the identification of thePKD1 gene,3 did not show any signs of HI (Dr I Cordeiro, Lisbon, Portugal, personal communication). Hence, the region between the breakpoint t(8;16) and thePKD1 gene may harbour a gene involved in HI. However, at present we cannot rule out that an unrelated genetic defect is underlying HI in the proband. Although clone 1.8F detects a transcript,9 it is not very likely that disruption of a gene by the breakpoint causes HI, as it was not noted in either the patient's father or his aunt. Alternatively, the HI observed in the index patient may be related to his partial trisomy of 8q.
This study is another example of genetic analysis initiated by careful clinical observations. In addition, it illustrates that chromosomal translocations must be considered as a cause of an autosomal dominant disease. Particularly when genes with a high mutation rate are involved, as is the case with the TSC genes, unaffected parents with multiple affected offspring are not uncommon. This can usually be attributed to gonadal or gonosomal mosaicism,22 which implies a relatively low recurrence risk and reduces the carrier risk of the parents' sibs to the population level. The involvement of a familial translocation, however, confers a 50% recurrence risk to the carrier parent, as well as an enhanced carrier risk to relatives.
Acknowledgments
We thank D van Allewijk, B Smit, B Beverloo, and A Hesseling-Janssen for providing us with the DrgI probe, the CMYC probe, the 8q terminal probe, and the PKD1/TSC2 probes, respectively, the technicians of the cytogenetic laboratory for the chromosome slides, and Professor H Galjaard for his continuous support.