Article Text

Abstract
Primary ciliary dyskinesia is an autosomal recessive condition characterised by chronic sinusitis, bronchiectasis, and subfertility. Situs inversus occurs in 50% of cases (Kartagener syndrome). It has an estimated incidence of 1 in 20 000 live births. The clinical phenotype is caused by defective ciliary function associated with a range of ultrastructural abnormalities including absent dynein arms, absent radial spokes, and disturbed ciliary orientation. The molecular genetic basis is unknown. A genome scan was performed in five Arabic families. Using GENEHUNTER, a maximal multipoint lod score (HLOD) of 4.4 was obtained on chromosome 19q13.3-qter at α (proportion of linked families) = 0.7. A 15 cM critical region is defined by recombinations at D19S572 and D19S218. These data provide significant evidence for a PCD locus on chromosome 19q and confirm locus heterogeneity.
- cilia
- Kartagener syndrome
- linkage
- 19q
Statistics from Altmetric.com
Primary ciliary dyskinesia (PCD, OMIM 244400) is an autosomal recessive disorder with an estimated incidence of 1 in 20 000 live births.1-3 The condition is characterised by recurrent sinopulmonary infections, bronchiectasis, and subfertility.1-3 Diagnosis is made by finding abnormal ciliary function on motility studies and abnormal ciliary structure on electron microscopy of brushings or biopsy of nasal mucosa.2 4 Disease severity varies from mildly affected subjects to severely disabled patients requiring heart and lung transplantation.1-3
The clinical phenotype is caused by dysmotility of the cilia which are complex structures composed of over 200 different polypeptides.5 6 Cilia are closely related structurally to the flagella of sperm and many protozoa (for example,Chlamydomonas). Microtubule protofilaments are arranged in the axoneme in the well known “9 + 2” pattern (fig1).7 Adjacent peripheral doublets are connected to each other by nexin links and to the inner pair by radial spokes. The peripheral doublets slide relative to each other in an ATPase dependent reaction involving the outer and inner dynein arms.8-11In humans, ciliated epithelium lines the respiratory tract including the sinuses and middle ear, the ependyma of the brain, the female oviduct, and the male vas deferens. It plays an important role in mucociliary clearance in the respiratory tract. A variety of ultrastructural abnormalities of cilia have been described in PCD. These include absent dynein arms (70-80% of cases), absent radial spokes (5-10%), microtubular transposition, and random ciliary orientation.12-14
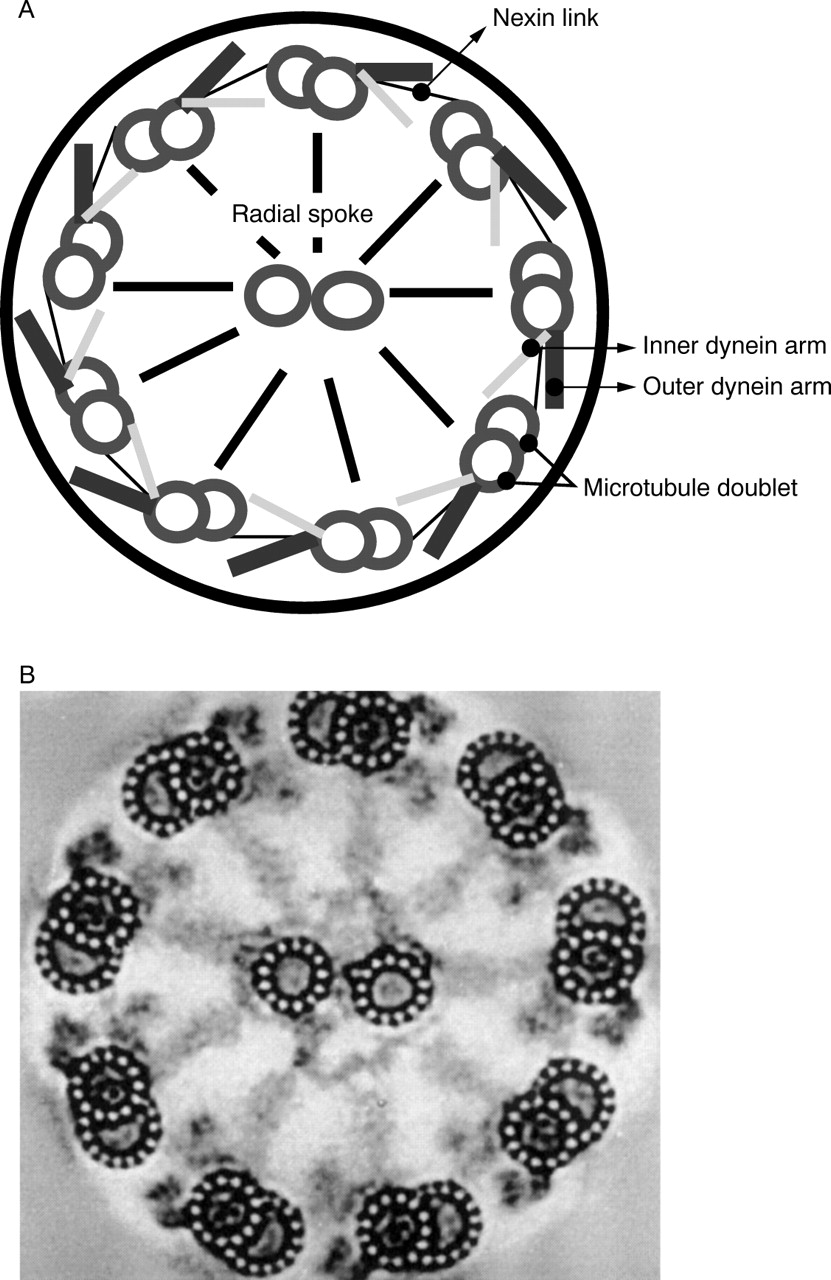
(A) A schematic representation of a ciliary axoneme illustrating the 9 + 2 microtubule arrangement and some of the microtubule associated proteins including the dynein arms. (B) A transverse section of a cilium as seen under transmission electron microscopy.
The molecular genetic basis of PCD is unknown. No gene or locus had been identified although linkage studies have been reported.15-17 About half of the patients have defects of laterality, usually situs inversus, a mirror image reversal of the left-right axis (LRA).18 This association is known as Kartagener syndrome19 and has been observed with all types of ultrastructural abnormalities except in the so called “microtubular transposition defect” and “overlong cilia”.18 The co-occurrence of ciliary dysfunction with situs inversus indicates that cilia play an important role in this highly conserved and complex pathway of LRA determination. However, the relationship between the ciliary defect and the abnormalities of laterality remains unknown. A number of hypotheses have been proposed, a common feature of which is that the primary defect resides in the cytoskeleton and that both ciliary motility and situs determination are influenced by the cytoskeletal defect.18 20 21 The only human gene unequivocally associated with situs abnormalities so far isZIC3, a zinc finger transcription factor, in which mutations have been identified in subjects with X linked and sporadic laterality defects.22 At present, three genes with a role in ciliary structure or assembly have been implicated in LRA determination: lrd (left-right dynein),hfh-4 (hepatocyte nuclear factor/forkhead homologue 4), and kif3B (kinesin member 3B).23-25
We have ascertained an extensive family resource and are undertaking genome searches to identify the genes responsible for PCD. Genetic heterogeneity is anticipated and families have therefore been grouped and analysed initially according to their population of origin or ultrastructural phenotype or both. We present the results of linkage analysis in five families of Arabic origin providing conclusive evidence for a PCD locus on chromosome 19q.
Subjects and methods
FAMILIES
The present report includes data from five families of Arabic origin, four from Saudi Arabia and one from Israel (family 89). DNA was available from a total of 32 subjects (12 affected, five of whom have situs inversus). The parents in all families are first cousins except in family 72 from Saudi Arabia. Diagnosis of affected subjects was made according to well established clinical criteria (table 1) supported by abnormal ciliary motility studies and electron microscopy (EM) of brushings or biopsy of nasal mucosa. An ultrastructural phenotype of absent outer dynein arms was shown in affected subjects where available.
Summary of diagnostic features of affected subjects
MARKER TYPING
Genomic DNA was extracted from white cells by standard methods. DNA was amplified by PCR using fluorescently labelled primers. PCR was performed in 96 well microtitre plates (Hybaid). Each well contained 30-60 ng of genomic DNA, 1.5-2 mmol/l MgCl2, 10 × reaction buffer (GIBCO), 200 mmol/l each of dATP, dTTP, dGTP, and dCTP, 50 ng of each primer, and 0.05 units of Taqpolymerase (GIBCO), in a total volume of 20 μl. Thirty cycles were performed in a thermocycler (Hybaid OmnigeneTM). Alleles were separated on a 6% polyacrylamide electrophoresis gel for three to four hours at a rate limiting voltage of 1000 V with a model 373XL DNA sequencer (Applied Biosystems). Analysis of the allele sizes was performed by GENESCANTM 672 (v 1.2) and Genotyper software (Applied Biosystems) using GENESCANTM -500 TAMRA as a size standard.
LINKAGE ANALYSIS
Linkage analysis was performed under the assumption of autosomal recessive inheritance with a penetrance of 0.9, disease allele frequency of 0.015, and a phenocopy rate of 0.001. Multipoint analysis was performed using the GENEHUNTER program.26 The marker loci from chromosome 19q that were included in this analysis were D19S572 - 4 cM - D19S418 - 3 cM - D19S605 - 5 cM - D19S404 - 2 cM -D19S214 - 1 cM - D19S218. Each marker was assumed to have five alleles of equal frequency. The relative positions of marker loci (fig 2) are based on recently published maps and chromosome 19 specific databases (Lawrence Livermore National Laboratory).27-29
{kind=link}
{kind=link}
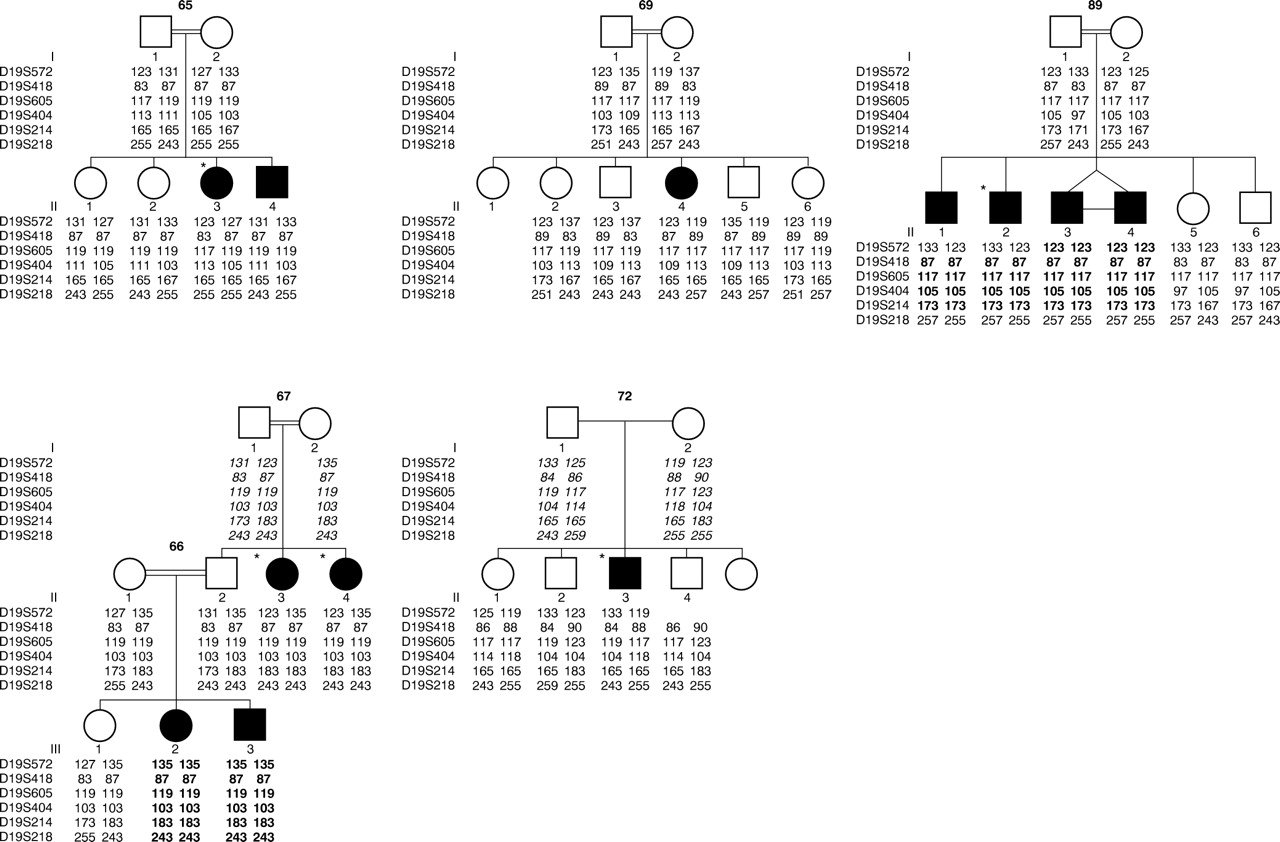
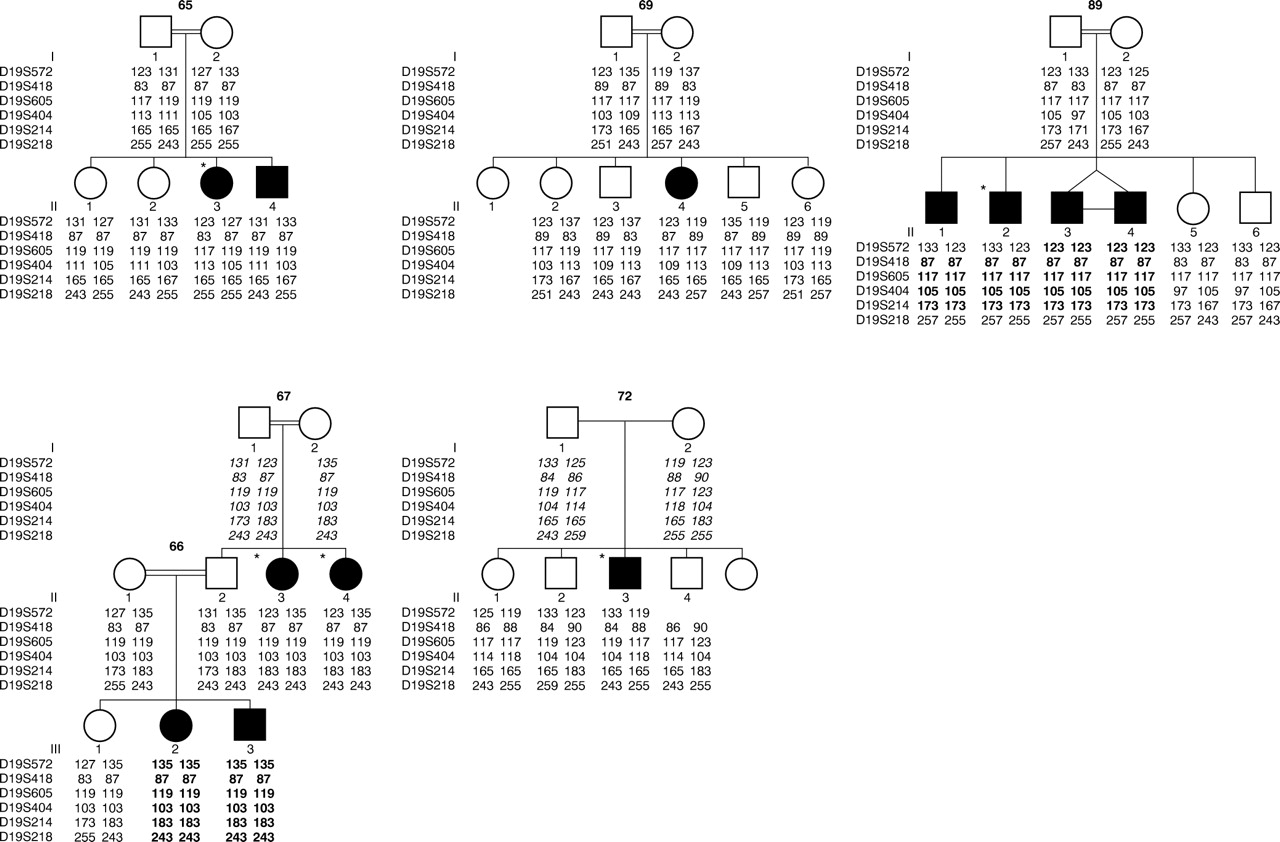
Pedigrees and chromosome 19q haplotypes. The genetic distance between D19S572 and D19S218 is approximately 15 cM (sex averaged). Regions of homozygosity by descent in affected subjects are highlighted in bold. Deduced marker data are shown in italics. Subjects with situs inversus are indicated by *.
Results
Over 300 microsatellite markers were typed and excess homozygosity was identified on chromosome 19q. Further analysis of high density mapping data using GENEHUNTER gave a maximum multipoint lod score (HLOD) of 4.4 at D19S404 and D19S218, allowing for heterogeneity, at α (proportion of linked families) = 0.7 on chromosome 19q13.3-qter. The HLOD remains over 4.0 from D19S572 to D19S214 with an α which varies only from 0.65 to 0.7. Haplotype analysis confirms linkage in families 66/67, 72, and 89 and non-linkage in families 65 and 69. Obligate recombinants at D19S572 and loss of homozygosity at D19S218 in family 89 define a critical region of about 15 cM. There is so far no evidence of linkage disequilibrium as no common haplotype or allele is present among affected subjects in the linked families. In family 65, the two affected sibs have inherited different parental chromosomes and in family 69 the affected subject shares common parental chromosomes with her unaffected sibs. These data therefore support non-linkage in these two families. The pedigrees and chromosome 19q haplotypes are shown in fig 2.
Discussion
This is the first report of the mapping of a locus for primary ciliary dyskinesia. Primary ciliary dyskinesia (PCD) is not an uncommon condition with an estimated incidence of about 1 in 20 000 live births. This is likely to be an underestimation because of the difficulties in making the diagnosis. Some of the clinical features such as otitis media, sinusitis, and recurrent pneumonia are common and are shared with other conditions including cystic fibrosis, immunodeficiency, and asthma. The presence of situs inversus obviously prompts consideration of the diagnosis and allows it to be made more confidently. Confirmation ideally requires nasal brushings/biopsy which is only available at specialised centres and is poorly tolerated by children. Disease severity also varies and some subjects may only manifest very minor or few symptoms.
The affected subjects in our families have been investigated appropriately (table 1). Investigations to exclude cystic fibrosis and immunodeficiency have been performed in most patients. The presence of bronchiectasis has been documented by x ray or CT scan. All families except 69 have at least one affected subject with situs inversus. The affection status can be firmly established on the available evidence from the clinical features and laboratory investigations. The parameters of our linkage analysis are appropriate, assuming a highly penetrant recessive model with a phenocopy rate of 0.001. The disease allele frequency of 0.015 was chosen to correlate with the estimated incidence.
Our data provide conclusive evidence for a PCD locus on chromosome 19q in a subset of these families. Genetic heterogeneity is expected in PCD in view of its wide range of ultrastructural phenotype. Grouping of families according to ultrastructural phenotype or population of origin is therefore a sensible strategy in a disease with expected genetic heterogeneity. All five families are Arabic in origin and affected subjects have a similar ultrastructural phenotype of absent outer dynein arms. Our linkage data support the presence of genetic heterogeneity of PCD even within these families of common population origin and ultrastructural phenotype. Work is in progress to examine for evidence of linkage in this region in other families/population groups, especially those with absent outer dynein arms.
The gene will be identified using a positional candidate cloning approach. The present 15 cM critical region is defined by recombinations at D19S572 and loss of homozygosity at D19S218 in family 89. This critical region will be further refined by investigation of meioses with flanking recombinants or identification of linkage disequilibrium. The extent of linkage disequilibrium depends on the existence of an ancestral disease mutation, the age of the mutation, and the degree of inbreeding. At present no evidence of linkage disequilibrium has been detected. High density marker typing in the linked families and the rest of the family resource with absent outer dynein arms at this locus is in progress.
Existing knowledge of the biology of cilia and of LRA determination provides a relatively firm foundation for the evaluation of transcripts as potential candidates in this region. Obvious candidates will include genes encoding structural components of cilia (for example, dyneins, tubulins, and microtubule associated proteins). Chromosome 19q13.3-qter is a gene rich region and contains a large cluster of zinc finger containing (ZNF) genes. This is of potential interest as mutations inZIC3, a zinc finger transcription factor, have been identified in subjects with X linked and sporadic laterality defects.22
The association of situs inversus with PCD (Kartagener syndrome) remains unexplained. These patients have an increased risk of cardiac malformations which confers significant morbidity. The cloning oflrd, an axonemal dynein heavy chain gene, in the inversus viscerum(iv) mouse support an important role for dyneins in the LRA determination pathway.23 More recently, work on nodal cilia in mice has provided strong evidence that cilia play a primary role in initiating this process. Nodal cilia are monocilia which are located in the primitive node in the normal embryo. They lack the two central microtubules and therefore adopt the 9 + 0 ultrastructural configuration. They have been shown to be motile and to create a directed flow of extraembryonic fluid which could produce a gradient of putative morphogen along the left-right axis in the node. Mice with targeted disruption of the kinesin family memberkif3b gene, which encodes a component of a microtubule plus end directed motor involved in intraciliary transportation, exhibit absence of nodal monocilia and randomisation of left-right asymmetry.25 It is therefore plausible that any defect producing abnormal cilia including the nodal cilia will result in PCD with randomisation of left-right asymmetry.
Determination of the molecular basis of PCD will provide information relevant both to clinical practice and basic biomedical science. New non-invasive approaches to diagnosis and carrier detection will become available and ultimately new methods of treatment should emerge. Gene therapy, for example, is a feasible long term option in a recessive disorder dominated by respiratory tract pathology. Moreover, it is possible that allelic variation at these loci causes a broader phenotype than presently recognised, including male subfertility or chronic secretory otitis media. The identification of the protein products encoded by PCD genes should provide new insights into the molecular mechanisms involved in the assembly and function of cilia and contribute to understanding of the pathway which determines the left-right axis in vertebrates.
Acknowledgments
This work was funded by Action Research. MM is a MRC Clinical Training Fellow.
References
Linked Articles
- Correction