Article Text

Abstract
Although trisomy of chromosome 21 is the most prevalent human genetic disorder, data from partial 21 aneuploidies are very scanty. Eight different partial aneuploidies for chromosome 21 were characterised by fluorescence quantitative PCR. Allelic dosage analysis was performed for each patient using 25 CHLC STRs covering the entire q arm. The length of the corresponding trisomies and monosomies was ascertained for five partial trisomics and three partial monosomics. All trisomic patients carried unbalanced translocations involving chromosome 21, whereas one of the monosomic patients bore a ring chromosome 21 and another showed an interstitial deletion of chromosome 21. The chromosomal breakpoints of two partial trisomy patients could be clearly delimited. However, the other three trisomies involved most of the 21 q arm as three allelic doses were detected for each marker. Although these latter patients do not show all the features of Down syndrome, genotype/phenotype correlations agree with previously reported data. The chromosomal breakpoints observed in two partially monosomic patients helped further to define the region involved in different phenotypic features associated with chromosome 21 monosomy. Telomeric material loss was also detected in a patient bearing a ring 21 chromosome. The parental origin of the aneuploidy was assigned for each case, which allowed us to conclude that two of the monosomic cases originated from de novo chromosomal rearrangements. There was no correlation with parental sex in contrast to trisomic patients originating from meiotic non-disjunction.
- Down syndrome
- partial trisomy
- partial monosomy
- chromosome 21
Statistics from Altmetric.com
Aneuploidies involving chromosome 21 are the most prevalent chromosomal abnormalities in humans and occur in approximately 1 in 700 births. A very low percentage of these aneuploidies are partial trisomies or monosomies, both difficult to detect as only a small region of chromosome 21 is involved. Prenatal diagnosis of these types of disorders used to be by conventional cytogenetics. Nowadays, FISH (fluorescence in situ hybridisation) allows the identification of most cases of aneuploidy, including small chromosomal translocations, otherwise undectectable by standard karyotyping. However, there are still a few cases, such as minute internal duplications, which remain unable to be identified through this approach and one major drawback of FISH is the quantity of amniotic nuclei needed for hybridisation to chromosomal probes, owing partly to amniocyte resistance to lysis.1
Rapid molecular methods involving the use of PCR (polymerase chain reaction) for prenatal diagnosis of chromosomal disorders have been reported.1-5 PCR allows amplification of a particular segment of DNA with simplicity and versatility. The highly polymorphic STRs (short tandem repeats or microsatellites) are the most useful tools to undertake these analyses. Although dinucleotide microsatellites show the highest heterozygosity indices and are the most frequent STRs, identification of the individual alleles can be blurred by the “mirror bands” produced by slippage of the DNA polymerase. Therefore, tri- and tetranucleotide microsatellites are the markers of choice as they constitute a compromise between heterozygosity, random distribution, and sharper resolution. Overall, they are more reliable for allelic dosage analysis. This is a critical point when dealing with aneuploidies, among which those affecting chromosome 21 are the most frequent.
The analysis of STR alleles in aneuploidies may render additional information if parental samples are available, as the parental origin and also the meiotic phase in which non-disjunction occurred can be easily determined.
Very few cases of partial chromosome 21 aneuploidy have been reported so far, but they have been crucial for genotype-phenotype correlation. Nonetheless, in order to confirm and refine this preliminary phenotypic map, additional information from other patients is needed. Data gathered by several research groups have been used by J M Delabar to construct a web accessible database of chromosome 21 partial aneuploidies, which constitutes a valuable reference for newly reported data (http://www.infobiogen.fr/services/aneu21/).
Using 25 STRs we have characterised the extent and boundaries of eight chromosome 21 rearrangements. We suggest that the set of markers described is suitable for use in prenatal diagnosis and in the characterisation of partial trisomies and other aneuploidies.
Methods
GENOMIC DNA ISOLATION
Peripheral blood was obtained from patients with different chromosome 21 abnormalities as well as their parents when available. Genomic DNA was extracted using a standard method.6 The phenotype of all probands was ascertained by local clinicians and their karyotypes had been previously analysed by conventional cytogenetics in each hospital (table 1 7 8). Blood samples were obtained from all the subjects after informed consent following the tenets of the Declaration of Helsinki.
Karyotype of probands
PCR CONDITIONS
The primers of the tri- and tetranucleotide STRs used were characterised by the CHLC (Cooperative Human Linkage Consortium) and GDB (Genome Database). D21S1261 was the only dinucleotide marker used in this analysis. An integrated map for their physical location is accessible in the web from NCBI, MIT, CHLC, and Généthon.
The forward oligonucleotide primers were 5′ end labelled with FAM. PCR amplification was performed in a total volume of 25 μl containing 30 ng of genomic DNA, 200 μmol/l dNTPs, 2-10 pmol of each primer, 1 × Dynazyme buffer (1.5 mmol/l MgCl2) except for markers D21S1993 and GATA148F04, in which MgCl2 was added to a final concentration of 2.5 mmol/l. After the first denaturation step of two minutes at 94°C, 1.5 U Dynazyme was added to the mixture. Two step PCRs (9600 Perkin Elmer, Applied Biosystems) were carried out for 25 cycles of 40 seconds at 94°C and 30 seconds at 52-58°C (the annealing temperature was adjusted in each case), followed by the final extension step of five minutes at 72°C.
PCR products (0.5-1 μl) were added to 0.5 μl of GeneScan 500 Rox marker and to 2.5 μl of 1:5 loading buffer-formamide mixture, resolved in a 377 ABI PRISM, and analysed using GeneScan analysis 2.0.2 software (Applied Biosystems), which assigned size and area to each allele. Three replicas were performed and analysed for each sample. We considered trisomy when three peaks at 1:1:1 ratio or two peaks at 2:1 ratio were observed. Quantitative analysis with a disomic autosomal STS (exon 5 of the antigen S gene, located in chromosome 2) was also performed as confirmation of trisomy for some markers. Ratios ranging from 1.3 to 1.8 were considered evidence for trisomy. Markers yielding lower ratios were considered disomic. Nonetheless, when using fluorescent primers, multiplex PCR with disomic and trisomic markers is not always successful owing to dye steric hindrance. Cases in which only one peak appeared were considered uninformative.
Results
Eight patients with different chromosome 21 abnormalities were analysed with a set of 25 STRs spanning the entire length of the 21 q arm (fig 1). The data for each microsatellite marker, arrayed from centromere to telomere, and the chromosomal region involved in the aneuploidy are shown in table 2.

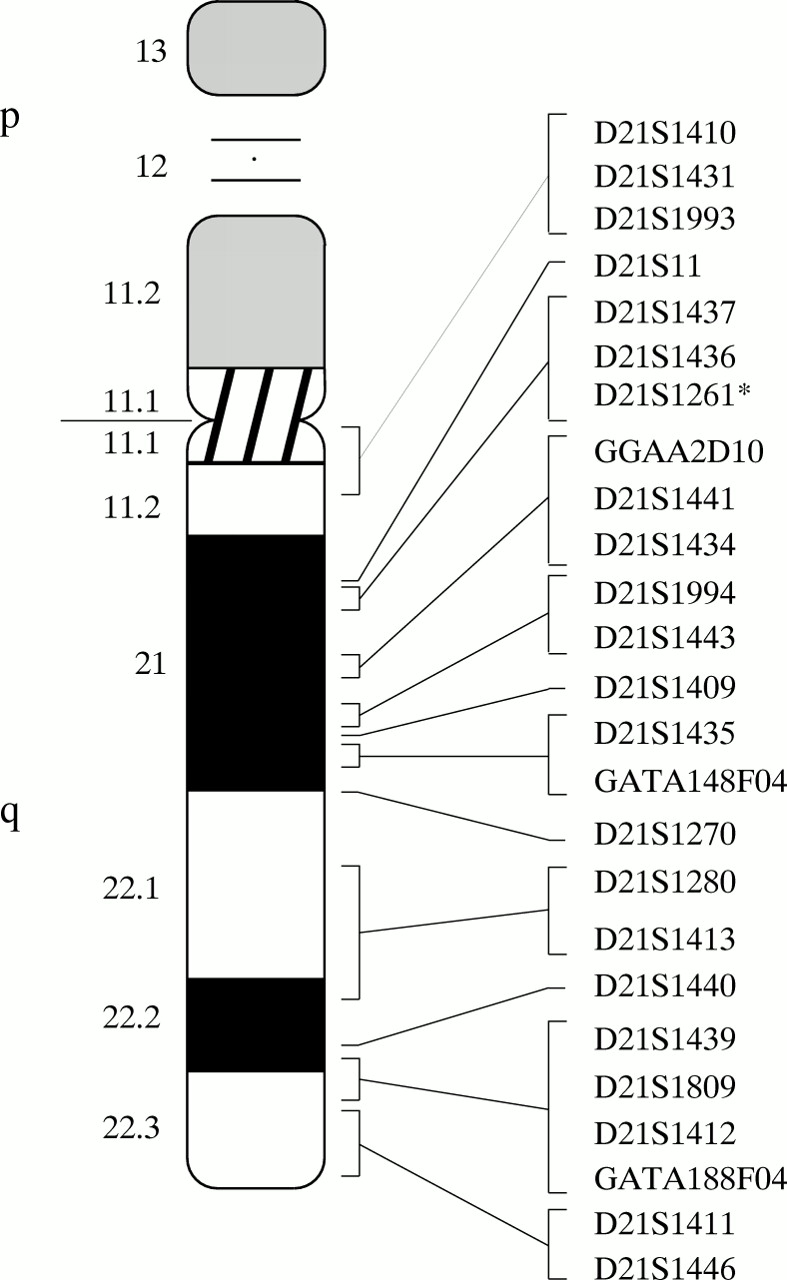{kind=link}
Location of the 25 CHLC and GDB markers used in this work, arrayed along the chromosome 21 q arm. *According to GDB, the D21S1261 dinucleotide marker used in this study has additional names, D21S1263 and D21S1417. Two other markers also have the same name, but they are tetranucleotide polymorphisms which use other amplimers and are located in other positions.
Allelic dosage analysis for the microsatellite markers used, arrayed from centromere to telomere
Tri- and tetranucleotides are highly polymorphic, so in most cases the trisomic nature of the sample could be established, side stepping the quantitative analysis, as three alleles at 1:1:1 ratio were detected. Two alleles detected at ratio 2:1 were considered as evidence for trisomy. In some cases, confirmatory quantitative analysis was performed by using a marker located in another (disomic) chromosome. When only one peak was amplified, quantitative PCR analysis was also performed. However, multiplex PCR when using fluorochrome labelled primers is not always successful and so these markers were considered uninformative.
Five probands were partial trisomics. Most of them had inherited an unbalanced gamete from a parent bearing a reciprocal translocation. In these cases, the correlation between genotype and phenotype was not straightforward and the clinical traits could not be entirely ascribed to the chromosome 21 trisomy. In most patients, almost the entire 21q arm was involved in the trisomy, as three allelic doses were detected for each marker analysed. When available, DNA samples from the parents were used to estimate the parental origin of the aneuploidy.
Proband ACR showed the smallest trisomy. The chromosome 21 breakpoint was located between markers D21S1280 and D21S1413, physically mapping within the 21q22.1 region. Therefore, the DSCR (Down syndrome critical region), involved in most Down syndrome (DS) patients,9was trisomic. The aneuploidy in this patient was the result of a rearrangement of the paternal chromosome 21.
The distal half of the 21 q arm was trisomic in proband EJL, as shown by the chromosomal breakpoint located between markers D21S1409 and D21S1435, mapped to 21q21.2-21.3 between D21S11 and the APP gene.
In probands SGH, EVC, and NMR the length of the partial trisomies could not be defined accurately as all the markers showed three allelic doses. Consequently, the chromosomal breakpoints must be located very close to the centromere.
Also, three probands partially monosomic for chromosome 21, CGM-10, CGM-14, and SPG, were included in this study. Proband CGM-14 showed a ring chromosome 21 with telomeric material loss. We detected a single allelic dose for markers D21S1411 and D21S1446, both mapping in the subtelomeric region of 21q22.3. Probands CGM-10 and SPG showed significant monosomy affecting the proximal 21 q arm. In both cases, their maternal chromosome bore a deletion from the centromere to 21q21.3.
Discussion
Recent data support the use of fluorescent quantitative PCR to analyse chromosomal abnormalities.1-5 The design of primers, the choice of an efficient fluorochrome (such as FAM), and the number of cycles restricted to the exponential phase of the amplification reaction (24 to 26) are critical factors for reliable quantification of the amplified products. In the present study, the PCR conditions for the 25 STR markers of the chromosome 21 have been optimised.
This approach offers some advantages over FISH; the parental origin of the aneuploidy can readily be assigned and the time required for the analysis is considerably reduced, as there is no need to establish cell cultures. Also, for prenatal diagnosis, (1) tests could easily be standardised using a few selected markers covering different chromosome 21 regions, (2) a large number of samples could be processed at once, (3) the assay is reliable even when few cells are available, and (4) results are gathered in a short time, allowing the parents to decide about the termination of a pregnancy without unnecessary delay. In fact, a very recent report5 shows the suitability of this approach when a small set of selected markers from chromosomes X and 21 is used. Although FISH might be the first choice for routine analysis of aneuploidies, partial aneuploidies may remain unnoticed unless different probes spanning all chromosomal regions are used. Also, molecular data allow the chromosomal breakpoints to be refined more quickly than with FISH and more accurately in comparison to conventional cytogenetics (see below the results of one of our patients). Knowledge of the parental origin of the aneuploidy could be important for genetic counselling and prenatal diagnosis especially when considering families where one of the parents carries a translocation and, thus, the descendants could inherit partial trisomies or monosomies. Moreover, fluorescent quantitative PCR is clearly advantageous over other techniques when dealing with submicroscopic rearrangements, such as interstitial deletions and small duplications, where a dense coverage of markers is needed.
The analysis of partial aneuploidies allows genotype and phenotype to be correlated so that the clinical features of Down syndrome (DS) can then be assigned to specific chromosomal sites. Although the genetic order and distance of tetranucleotide markers is well established, their physical location has not been mapped accurately and awaits further refinement. This hampers the comparison of our data with those reported by other authors, who mainly used dinucleotide and STS markers.
PARTIAL CHROMOSOME 21 TRISOMIES
Most of the DS traits appear when the DSCR region (located around the D21S55 marker and roughly comprising from distal 21q22.1 to proximal 21q22.3) is trisomic.9 10 In our patients, most of the 21q arm is involved in the trisomy, including the DSCR. It is therefore understandable that these patients display most of the phenotypic features associated with Down syndrome. The translocations affected different chromosomes in each case, and no traits other than those of DS have been reported. Thus, the effects of the unbalanced monosomies may match, overlap, or be masked by the DS phenotype. Clinical assessment of the phenotypic features is shown in table 3. Unfortunately, not all the traits on the list of Jacksonet al 11 could be assessed in all the patients.
Phenotypic features of the patients with different chromosome 21 aneuploidies. (Proband CGM-14 has not been included as his phenotypic clinical traits do not coincide with those considered in Jackson’s list: see text)
We have compared the clinical and molecular data from our patients to those previously reported.9 10 Proband ACR suffers from a severe congenital heart defect, which has required several surgical interventions. As the chromosome 21 breakpoint in this patient maps within the region responsible for Down syndrome congenital heart disease, DS-CHD,12 our results are consistent with the assigned position of the gene or genes causing this pathology. Probands SGH and NMR, with a partial trisomy involving nearly all the 21 q arm, do not have the typical transverse palmar crease, whereas other patients, with a more restricted trisomy, have this trait (see probands EJL and ACR). Probands SGH and EVC do not have microcephaly, in contrast to proband NMR, who bears the same trisomic region. Also, probands EJL, ACR, and EVC do not have Brushfield spots.
Interestingly, molecular data can help to refine chromosomal breakpoints first approached by cytogenetic analysis. Such is the case of patient SGH, a proband with an unbalanced translocation involving chromosomes 19 and 21. Previous cytogenetic interpretation of the G banded karyotype located the chromosomal breakpoints in 21q21 and 19q13.3 (table 1 7), so that the proximal region of chromosome 21 was assumed to be disomic. However, according to our molecular analysis, this region had to be trisomic, and thus included in the translocation, because markers on 21q11.1 and 21q11.2 were all present in three allelic doses. This apparent inconsistency could be reconciled when considering that the negative G bands 19q13.3 and 21q11.2 look very similar and could be easily mistaken for each other in a cytogenetic analysis. Therefore, refinement through molecular data indicates that the breakpoints are located in 19q13.2 and 21q11.1/21q11.2.
Variability in the observed DS phenotype is common. In our case, proband ACR, bearing the smallest partial trisomy, shows most of the phenotypic traits associated with DS while these pathological features are absent in other probands with longer trisomic regions, such as EVC and NMR. Various explanations could account for this apparent contradiction: (1) other genetic or epigenetic factors may modify the penetrance of a particular trait, (2) recessive and dominant alleles may contribute differently to the phenotype depending on the allelic dose ratio (AAA, aaa, Aaa, AAa v AA, aa, Aa), (3) the phenotypic effects of the unbalanced monosomies caused by the chromosome 21 translocations are unknown, and (4) clinical assessment is subject to bias. Overall, the uniqueness of each case makes it very difficult to evaluate each of these factors.
PARTIAL CHROMOSOME 21 MONOSOMIES
Partial monosomies of chromosome 21 are not as frequent as partial trisomies. Ring chromosomes account for most of the reported cases of monosomy while deletions covering a larger region are scarce. The high degree of phenotypic variability in patients with similar deletions makes it difficult to assess the phenotypic influence of a dominant or a recessive allele in hemizygosity.
Proband CGM-14, a male, shows a ring chromosome 21 involving telomeric loss. His clinical features (not listed in table 3) are hypoplasia, agenesis of the 5th finger, EEG anomalies, and extended subcortical brain atrophy, as detected by CT scan. According to reported data, the phenotype associated with patients bearing a ring chromosome 21 is variable and may depend on the length of the telomeric deletion or even the sex of the patient.13 Some patients do not show clinical anomalies other than azoospermia and infertility (in males) and increased risk of miscarriages and bearing children affected by Down syndrome (in females),14-16 whereas others have mental retardation,13 microcephaly, hypertelorism, downward slanting palpebral fissures, and psychomotor retardation.17 Some authors claim that monosomy of the juxtatelomeric region (the most distal 1 Mb) in the ring chromosomes 21 has very little or no phenotypic effect, particularly when considering mental retardation.18-20 However, the phenotypic variability among ring 21 probands cannot be ascribed only to mere differences in the length of the deleted chromosomal regions, as it has also been described in families segregating a stable ring chromosome 21.21 Thus, some other as yet unknown genetic or epigenetic factors must be involved.
Probands CGM-10 and SPG show monosomy of the proximal half of the 21q arm, spanning from the centromere to 21q21.2-21.3. Although SPG bears a longer deletion (as detected by marker D21S1443), the physical location of their chromosomal breakpoints is relatively close. Common clinical traits to both patients are epicanthic folds and flat nasal bridge, although others, such as short stature, microcephaly, high arched palate, or hypospadias, have been clearly determined for only one of the patients. Also, CGM-10 shows abnormal dermatoglyphs and mental retardation while SPG has brachycephaly, downward slanting palpebral fissures, and a mild heart defect. Again, a very variable phenotype for monosomy 21 has been reported ranging from non-intellectual disability and mild clinical traits22 to facial dysmorphic features and mental retardation.20 Allegedly, differences in the length of the deletion would account for this phenotypic variability, the region spanning from APP toSOD1 being the critical region responsible for most 21 monosomy associated traits.20 23 The region deleted in CGM-10 includes neither APP norSOD1 whereas that of SPG comprises theAPP but not theSOD1 gene. In proband SPG, the marker GATA148F04, which is clearly disomic, is located distally but very close to D21S1435 (known to be located in YAC 922A3ceph, which contains the APP gene23). The genotype/phenotype correlation deduced from both patients is consistent with that reported by other authors.20 The features of epicanthic folds and flat nasal bridge map in the proximal half of the q arm, in the region deleted in the two probands. The trait of downward slanting palpebral fissures has been previously mapped by other authors to the region between marker D21S11 andAPP.23 Our data further place its location between markers D21S1994 (located in YAC 944f8) and GATA148F04 (located close to APP, see above). The gene (or genes) responsible for high arched palate has been previously mapped between APP andSOD1 23; according to our data it should be located very close toAPP.
Interestingly, many of the features associated with chromosome 21 monosomy (such as microcephaly, short neck, epicanthic folds, flat nasal bridge, low set/malformed ears, highly arched palate, and transverse palmar crease20 22) are also present in trisomic patients (DS). This concurrence favours the hypothesis that an excess or a deficiency in gene dosage may result in similar phenotypes. As already pointed out, the effects of recessive alleles are difficult to evaluate in a trisomy or monosomy (“Aaa” and “a” allele combinations may produce the same phenotypic trait in contrast to “Aa” or “AA” disomic combinations). This could well apply to genes involved in morphological traits, whose products interact at particular developmental stages. These genes are usually very finely regulated in their expression, both temporally and spatially. The variation of expression levels owing to three or one gene copies instead of two may deregulate other genes in a cascade effect. Nonetheless, this is a very controversial hypothesis as, for some features, opposite phenotypes appear in trisomic and monosomic patients, for example, downward slanting versus upward slanting palpebral fissures. However, according to our results, together with those of Korenberg et al,22these two phenotypic traits are caused by genes located in different regions of chromosome 21, providing further molecular evidence against the type-contretype theory of aneuploid phenotypes, as discussed by other authors.22
PARENTAL ORIGIN OF THE ANEUPLOIDY
STR analysis has allowed us to determine the parental origin of the aneuploidy (table 4). Most chromosome 21 trisomies are produced by maternal meiotic non-disjunction.24 However, partial trisomies are caused by a completely different mechanism, as they usually come from unbalanced gametes bearing a translocation. Probands SGH, EJL, and ACR have trisomies of paternal origin whereas probands EVC and NMR are maternal. Monosomic patients CGM-14 and SPG are of paternal and maternal origin, respectively. As their parents do not show the corresponding monosomies, these aneuploides originated from de novo deletions. In contrast, CGM-10 shows two different aneuploidies, partial trisomy of chromosome 13 and partial monosomy of chromosome 21, as he received an unbalanced gamete from a mother bearing a reciprocal translocation affecting these chromosomes.
Parental origin of the aneuploidy
Acknowledgments
We thank the patients and their families for their cooperation in providing blood samples. We also thank the Serveis Científico-Tècnics de la Universitat de Barcelona for the use of the 377 ABI PRISM and Robin Rycroft for revising the English. R Valero is in receipt of a fellowship from the CIRIT (Generalitat de Catalunya). This study was funded by Fundació Síndrome de Down/Marató de TV3-1993 and CICYT SAF96-0329.