Article Text

Abstract
We have reviewed published reports on patients with segmental aneusomy for chromosome 1p36 to help geneticists and other health professionals in the recognition of this emerging chromosomal syndrome. Terminal deletions of the short arm of chromosome 1 are associated with hypotonia and developmental delay (usually severe), growth abnormalities (growth retardation, microcephaly, obesity), and craniofacial dysmorphism with a large anterior fontanelle, prominent forehead, deep set eyes, flat nasal bridge and midface hypoplasia, ear asymmetry, a pointed chin, and orofacial clefting. Minor cardiac malformations, cardiomyopathy, seizures, and ventricular dilatation are the more common additional findings. Sensorineural hearing loss and variable ophthalmological anomalies have also been frequently observed.
Although the deletions can be detected by high resolution cytogenetic studies, confirmation by fluorescence in situ hybridisation is required in most cases. The majority of deletions are maternally derived. Molecular characterisation of 1p36 deletions has been undertaken in several cases, and it is likely that this condition is a contiguous gene deletion syndrome.
- monosomy 1p36
- contiguous gene deletion syndrome
Statistics from Altmetric.com
Two of the most important aims behind the characterisation of a “chromosomal syndrome” are to aid recognition of the phenotype and to document developmental progress. Until recently, there were few published reports of patients with “pure” monosomy for terminal chromosome 1p36. We provide a review of the published cases with 1p36 deletions and with interstitial deletions involving 1p36, in order to assist geneticists and other health professionals with the identification of patients and counselling of families with this chromosome abnormality.
Reported cases
Published cases of 39 patients with pure 1p36 monosomy are listed in table 1 together with karyotype, age at presentation, and reason for presentation.1-19 One case with mosaicism for monosomy 1p36 has been included.13 Five patients with monosomy 1p36.31-33 arising from unbalanced segregants have also been included in table 1, as the phenotypic effects from the derivative chromosome 1 can be attributed mainly to 1p monosomy.16-19 The dysmorphic features and malformations associated with pure lp36 monosomy are summarised in table 2 and discussed in the text. The incidence of dysmorphic features and malformations has not been given as a percentage because of differences in the completeness of the case reports. The deletion size of 17 additional patients has been reported, but because their clinical features were not listed, they are not included in this review.20 Photographs of a collection of typical patients with 1p36 monosomy are shown in fig 1. Cases with terminal 1p36 monosomy reported only in abstracts,1-3 5 7 two patients with interstitial deletions,14 15 and six patients with 1p monosomy resulting from the segregation of translocations,9 16-19who have possible additional chromosome imbalance, have been excluded from table 2.
Reported cases with monosomy for chromosome band 1p36
Clinical features of reported cases with monosomy for chromosome 1p36

Six children with 1p36 deletions, whose deletion sizes have been characterised.20 The patient numbers correspond to patient assignments reported in Wu et al.20 (A) Patient 6 at 5 years of age. (B) Patient 10 at 19 months of age. (C) Patient 12 at 13 months of age. (D) Patient 15 at 9 years of age. (E) Patient 23 at 31 months of age. (F) Patient 28 at 24 months of age. (All photographs reproduced with permission.)
Aetiology and genetics
The incidence of de novo monosomy for 1p36 has been estimated at 1/10 000 newborns.9 Underascertainment in the past has been attributed to difficulty in the detection of deletions involving the pale staining end of the chromosome with G banding.
The sex of the “deleted” patient was published in 35 cases; 13 males and 22 females have been reported (table 1). Several cases have been found at amniocentesis.12 21 Interestingly, in a retrospective analysis of 30 patients, five out of eight mothers who underwent prenatal serum screening during pregnancy had raised levels of maternal serum alphafetoprotein.21 The age at diagnosis in the remaining patients has ranged from the newborn period to 47 years of age. Thirteen patients presented in the newborn period or first month of life and all but five patients were younger than 10 years of age at diagnosis. The most common reasons for chromosome analysis were developmental delay, dysmorphism, seizures, and the investigation of multiple congenital anomalies, although one child was karyotyped because of congestive cardiac failure in the newborn period4 and two others were referred with possible Prader-Willi syndrome.9 13 Survival to adult life has been described6 and all but four children were alive at the time of reporting (table 1).
The breakpoints of the deletions have ranged from 1p36.13 to 1p36.33 with the majority occurring in band 1p36.2 (table 1). Successful detection has been accomplished by high resolution G banding.13 14 One study detected two deletions by analysis at the 850-1000 band level3 and another used a 600-800 band level.9 In most cases the deletions were confirmed with FISH using probe D1Z21 6 7 10 11 13 18 19 23 or using both probe D1Z2 and probe p58.9 20 Both FISH probes should be used to confirm the diagnosis, since some patients have been described who have been deleted for only one of these two probes.9 20Three deletions were ascertained during screening studies for submicroscopic chromosome deletions on panels of children with idiopathic mental retardation.8 10 11 These findings lend support to the high estimated occurrence of this deletion syndrome.9
In the largest study of “pure” monosomy 1p36, 21 out of 27 de novo deletions were maternally derived.20 The deletions ranged in size from the “minimal” deletion interval (includes marker D1S243 and probe p58) to the largest deletion, with a proximal breakpoint near marker D1S2736.20 Three other cases have also been characterised molecularly and have shown breakpoints within this range.8 10 11 Thus the breakpoints are highly variable within 1p36. Molecular characterisation of deletions, along with phenotypic correlations, have yielded regions in which to search for genes for specific features of the syndrome which has resulted in the conclusion that the 1p36 deletion syndrome is likely to be a contiguous gene deletion syndrome.20 22
Clinical features
DEVELOPMENT
Developmental delay is almost invariable, although the small number of older children and adults with pure 1p36 monosomy reported has prevented a comprehensive assessment of adult potential. Four out of six children in whom IQ could be assessed scored <60 with speech more severely affected than motor development.9 One patient with an IQ <20 was unable to walk unassisted in her forties.6 Development was also described as severely delayed in five out of six patients in one study,7 in a 12 month old girl,18 in a 4 year old girl,17 and in a 16 year old boy.8 One girl was unable to sit independently at the age of 6 years10 and two others had no language acquisition at over 4 years of age.11 13 In contrast, three patients with the smallest documented deletions had complex speech abilities and mild mental retardation.20 24 However, it seems reasonable to conclude that intellect is severely compromised in the majority of patients.
GROWTH
Postnatal growth retardation was found in eight out of eight patients and microcephaly in four out of nine children in one patient group.9 In these patients, measurements at birth were normal, but reduced growth was detected during or after the first year of life.9 Pre- and postnatal growth retardation, with or without microcephaly, has also been noted in a number of other cases.1-4 6 8 10 11 15-17 19 Growth retardation in 1p monosomy can be severe10 and it is interesting that several patients with ring 1 chromosomes have also had extremely short stature.25-28
Paradoxically, children with obesity and hyperphagia resembling Prader-Willi syndrome have also been reported with terminal 1p deletions.2 4 7 9 13 This observation led to the characterisation of two separate phenotypes for 1p36 monosomy.2 4 7 However, it has since been recognised that the division of patients with 1p36 deletions into two groups on the basis of growth parameters is unhelpful in predicting other clinical features.9 The differences in growth were previously suggested to arise from variations in deletion size or imprinting,4 but no evidence of a correlation between growth parameters and deletion size or parental origin of the deletion has subsequently been found.9
Early puberty has been described in two female children, although their ultimate adult heights are predicted to be short.9Conversely, one boy with microcephaly had small genitalia and no signs of puberty at 16 years of age8 and the only reported adult female patient had growth retardation, small breasts, and sparse axillary and pubic hair.6
DYSMORPHISM
The most common dysmorphic features included a large anterior fontanelle,2 4 8-10 19 prominent forehead (also described as bossed or broad/long),4 6 10 11 18 19deep set eyes,6 9 13 17 19 and short, narrow, or slanting palpebral fissures.1 2 4 6 9-11 A flat nasal bridge2 4 9 15 17 and midface hypoplasia6 8 13 were also frequent. Other facial features were dysplastic ears or small, low set ears,1 2 4 6 9 10 12 13 16-19 a small mouth,4 8 11 13 19 or a mouth with downturned corners,6 11 13 19 and a pointed or prominent chin.6 9 10 Several children have had a long philtrum.10 11 Minor digital anomalies such as short fifth fingers or fifth finger clinodactyly,2 4 9 17camptodactyly,11 17 and small hands and feet9 11 13 were noted in some patients. Facial asymmetry4 8 10 11 and asymmetrical ears9 11 were common.
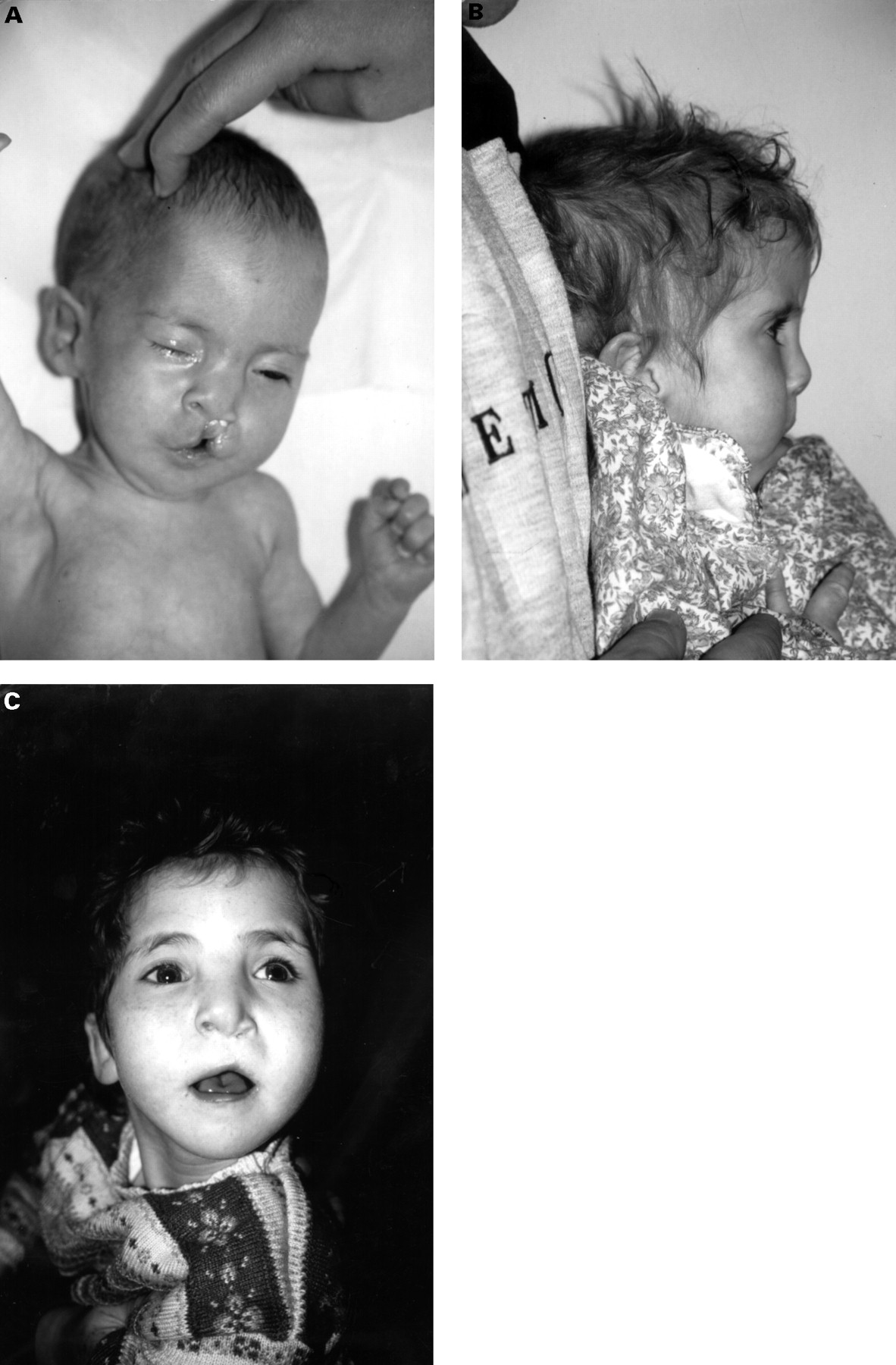
Orofacial clefting has been described in 10-40% of patients with 1p36 monosomy.2 4 9 In addition to reports of cleft lip/palate2 4 7 9 10 17 18 there are reports of one patient with a submucous cleft,3 five patients with a high arched palate,6 12-15 one boy with a flat, ridged palate,8 and a girl with prominent lateral palatine ridges.11 Examples of the facial appearance of a child with 1p monosomy, cleft lip, and cleft palate are shown in fig2.
{kind=link}
{kind=link}
Child with 1p36.2 deletion in the newborn period (A), profile at 3 years of age (B), and front view at 6 years of age (C).10
CARDIOVASCULAR SYSTEM
Four children have had infantile dilated cardiomyopathy and one of the four, who also had a patent ductus, had a degree of left ventricular failure greater than expected from the cardiac malformation.4 9 In two of these cases, the cardiomyopathies resolved by 9 months and 2 years of age,4and the cardiomyopathy was improving in the other two patients who have been on treatment with digoxin. Another child with a moderate sized atrial septal defect, tricuspid incompetence, and a small patent ductus arteriosus had left ventricular failure requiring medication for the first six months of life.10 Cardiac abnormalities were found in nine other patients and included tetralogy of Fallot,1 a small ventricular septal defect,15infundibular stenosis of the right ventricle,17 a patent ductus arteriosus,9 16 mild left pulmonary artery branch stenosis,9 and Ebstein anomaly with a patent ductus arteriosus.11 12 One case with an interstitial deletion of 1p36 had pulmonary atresia and a ventricular septal defect.14
CENTRAL NERVOUS SYSTEM
Hypotonia has been consistently documented, and in most cases was noted at birth.2-4 7 9-11 13 15 17-19 Epilepsy has also been found in the majority of children2-4 6 8-11 19 and myoclonic seizures,4 9 generalised tonic-clonic seizures,11 19 right sided focal fits,8simple and complex partial seizures,9 and infantile spasms9 have been described. In some children, seizures started in infancy and ceased in the first few years of life,3 9 whereas others have had persistent convulsions requiring long term medication.3 6 8-11 19 EEG abnormalities may be absent in children with transient seizures.9 A hemispherectomy was performed in one child for management of seizures.3
Cranial imaging has documented cerebral atrophy, ventricular asymmetry, and ventricular enlargement.9-12 16 17Hydrocephalus has occurred in two children.4 18 One report found hyperreflexia to be a common finding.2Disturbed behaviour has been described in some cases, including temper outbursts, banging or throwing objects, striking people, screaming episodes, and self-injurious behaviour (wrist biting, head striking).6 9 13 Autistic behaviour has also been noted.3 9 19
OPHTHALMOLOGICAL AND AUDITORY SYSTEMS
In one study, visual abnormalities were found in six out of eight patients and consisted of strabismus, sixth nerve palsy, amblyopia, refractive errors, anomalous optic discs, and lacrimal defects.9 Bilateral cataracts,6 iris colobomas,4 and moderate bilateral optic atrophy or pale optic discs8 18 have also been described. Rotatory nystagmus was found in three children3 8 18 and several others have had strabismus.4 10 13 18 Reduced visual acuity or refractive errors have been confirmed in other patients.10 15 19
Sensorineural or conductive hearing loss have been reported to be quite frequent. In the largest study,29 8/18 patients had bilateral high frequency sensorineural hearing loss (HFSNHL), 2/18 patients had severe conductive hearing loss, and 1/18 had mild HFSNHL, but had PE tubes in place at the time of the study. Other cases of hearing impairment have also been reported.6 10 13 18 19
MUSCULOSKELETAL SYSTEM
Kyphoscoliosis has been described in two cases and surgery was necessary in one child.6 8 A thoracic kyphosis secondary to muscular hypotonia and mild scoliosis has been mentioned in other reports.10 11 16 Contractures of the fingers and hips,1 dysplasia of the hip joints,18metatarsus adductus,2 and two patients with 11 pairs of ribs10 12 and a bifid rib10 have also been noted. Mild hemihypertrophy was found in one child.4
GENITOURINARY SYSTEM
Cryptorchidism has been found in two patients9 15and shawl scrotum,3 small genitalia,8 scrotal hypoplasia,9 and dilatation of the renal collecting systems10 have each been described in individual cases. No structural renal anomalies have been noted.
OTHER FEATURES
Anomalous findings reported in only one case were imperforate anus with rectovaginal fistula,1 abnormal pulmonary lobulation,19 congenital spinal stenosis,6polydactyly,4 and Sturge-Weber sydrome.3 One child with an interstitial deletion had a hairy skin patch14 and another had a telangiectatic skin lesion on her mid forehead and hyperpigmented macules.4
Monosomy 1p with additional chromosome imbalance
There are at least 10 reports of monosomy 1p and additional chromosome material arising from either unbalanced segregation of a chromosome translocation or as a de novo sporadic event.6 9 30-37 Because the additional chromosomal imbalance may be subtle, chromosome or FISH analyses should be performed on the parents of all 1p36 deletion syndrome patients. In the largest review,20 1 in 30 patients arose from unbalanced segregation of a parental balanced translocation. These reports of the clinical features in cases with double segment imbalance strengthen the association of malformations with 1p36 monosomy, although a contributory role from the additional chromosome imbalance cannot be excluded.
Cardiomegaly was observed in a patient with monosomy for 1p36-1pter and trisomy for 9p12-9pter.30 An infant with monosomy for 1p36.2-1pter had atrial and ventricular septal defects, seizures, and cerebral ventriculomegaly, although this child also had a duplication of 4q31.2-4qter.34 An infant and a 20 week old fetus with monosomy 1p36 and trisomy for the distal long arm of chromosome 20 were described with congenital heart disease (tetralogy of Fallot, truncus arteriosus, and ventricular septal defect), ophthalmological abnomalities (microphthalmia, optic nerve colobomas, foveal hypoplasia), and hydrocephalus.35
Two patients with monosomy for 1p36.31-1pter and trisomy for 19q13.42-19qter had seizures and one had optic atrophy, deafness, and strabismus.6 A man with seizures and a marked scoliosis had a translocation of Y chromosome material to chromosome 1 resulting in monosomy for 1p36.33 Megacystis with early urethral obstruction was found in a 15 week fetus with monosomy for 1p36-1pter and a duplication of terminal 10p.36
Terminal deletions involving 1p36 and extending proximally to 1p35,38 1p34,39 or 1p3340 have not been included in this review because of the difficulty in attributing clinical features specifically to band 1p36. However, it is interesting that one 9 year old child with a deletion of 1p35 was karyotyped for suspected Prader-Willi syndrome.38
Monosomy 1p and tumours of neural crest origin
There is strong evidence that one or more tumour suppressor genes map to 1p36.1-1p36.3 and are involved in the development of neuroblastoma and other tumours of neural crest origin.41-44 Loss of heterozygosity studies and microsatellite markers have been used to map the critical region for a neuroblastoma tumour suppressor gene to between D1S244 and D1S214.45 46 One child with an interstitial deletion of 1p36 was diagnosed with neuroblastoma at the age of 5 months15 and two other children who had balanced translocations with breakpoints at 1p36 have also developed neuroblastomas.47 48 In the largest study reported, no child with 1p36 monosomy (ages 6 months to 17 years) has as yet developed this type of tumour.20 It is possible that imprinting may be important in neuroblastoma formation49since one study showed that 16/17 allelic losses in this type of tumour were maternal in origin.50 In a study of 1p36 deletions, the three cases with the largest deletions in which parental origin could be determined had segmental aneusomy caused by a paternal deletion.20 If the tumour suppressor locus was proximally located within the deleted region of these patients, then the absence of neuroblastomas may be explained by the loss of the paternal, rather than the maternal allele.20 However, if the gene maps more distally in 1p36, then the finding of both maternally and paternally derived deletions in the patients fails to support a role for imprinting in neuroblastomas.20
Conclusion
We have reviewed 39 patients with “pure” 1p monosomy from medical publications in order to characterise the phenotype associated with this chromosomal syndrome. Terminal 1p deletions result in developmental delay (usually severe), abnormalities of growth (including growth retardation, microcephaly, or obesity similar to Prader-Willi syndrome), and dysmorphism with a large anterior fontanelle, prominent forehead, deep set eyes, flat nasal bridge and midface hypoplasia, ear asymmetry in shape or positioning, a pointed chin, and fifth finger clinodactyly/shortening. Minor cardiac malformations/cardiomyopathy, minor cerebral abnormalities, hypotonia, seizures, sensorineural hearing loss, and ophthalmological anomalies have been among the most frequently observed additional findings.
The variable nature of features that occur in patients with monosomy 1p36 necessitates monitoring for all the major complications. Medical management should include an echocardiogram, a hearing evaluation by brainstem evoked response testing, an ophthalmological evaluation, as well as the involvement of neurologists, cardiologists, nutritionists, otolaryngologists, cleft lip/palate specialists, speech pathologists, and physical and occupational therapists, to deal with the more common manifestations of this syndrome. A determination of the extent of the deletion at the molecular level can also yield important prognostic information. Prognosis and management decisions will also be augmented by future identification of candidate genes that, when deleted, result in the various phenotypic aspects of the syndrome.
Acknowledgments
We are most grateful to Professor Schinzel for kindly supplying details of his patient and to Dr Jill Clayton-Smith for reading the manuscript.