Article Text

Abstract
Recently five patients with an Albright hereditary osteodystrophy (AHO)-like phenotype were reported to have a subtelomeric deletion of the long arm of chromosome 2. These patients showed a striking resemblance to a number of patients from a large pedigree known to us for a long time. After molecular confirmation of a subtelomeric deletion in one patient, FISH analysis was used and a cryptic translocation between the long arms of chromosomes 2 and 8, t(2;8)(q37.3;q24.3), was detected. Remarkably, five proven and 10 probable cases with a 2qter deletion were found in the family, but none with an 8qter deletion. This was not explained by increased fetal loss.
The major clinical characteristics of terminal 2q deletion are a short, stocky build, round face, sparse hair, deeply set eyes, bulbous nose, thin vermilion border, brachymetaphalangism, seizures, and developmental delay. A specific behavioural phenotype consisting of periods of hyperkinesia and aggression can develop with age. The overall phenotype is sufficiently characteristic to allow clinical recognition.
The cytogenetic and molecular studies did not narrow down the common deleted region. Both testing of additional 2q markers and characterisation of other AHO-like patients with 2q37 microdeletions may help to define the candidate gene region.
- cryptic translocation
- t(2;8)
- 2q deletion
- AHO-like phenotype
Statistics from Altmetric.com
Clinical geneticists are regularly confronted with patients in whom clinical features or the pedigree suggest a chromosome abnormality, but in whom cytogenetic analysis fails to show an unbalanced karyotype, even on high resolution banding. In such cases, a submicroscopic chromosome abnormality can be present. Fluorescence in situ hybridisation (FISH) studies may be very helpful in this respect.
In 1968, a member of such a family was referred to our centre, followed by numerous others. These family members all showed mental retardation in combination with skeletal abnormalities. At that time the diagnosis of Albright hereditary osteodystrophy (AHO) was made (see Wilsonet al 1 for review). After more specific investigations, the patients were considered to have pseudopseudohypoparathyroidism (PPHP). In 1980 it became known that Gsα activity was reduced in patients with PHP and PPHP.2 3 Measurement of Gsα in one patient in this family, however, failed to show any abnormalities, raising doubts regarding the diagnosis. When Wilson et al 4 recently described five patients with an AHO-like phenotype, including a normal Gsα activity, and with subtelomeric deletions of the long arm of chromosome 2, the resemblance to the phenotype and history of our family was striking.
Here, we report on the results of molecular testing of one of our patients in whom molecular studies showed a terminal 2q deletion and on the clinical findings and cytogenetic studies using fluorescence in situ hybridisation (FISH) in the extended family. A detailed clinical description of the patients will hopefully contribute to the spectrum of the AHO-like phenotype.
Patients
The pedigree of the family under study is presented in fig1.

Pedigree of the present family.
No family member is known to have had recurrent miscarriages. Six out of 13 children of II.1 died shortly after birth (III.7-12). On four of them there is no information; one died at 6 weeks and one at 8 days, both of unknown causes.
INDEX CASE (V.10)
Patient V.10 was not the first patient seen in this family, but as her sister asked for genetic counselling, and at that time the definitive diagnosis was made, we regard her as the index patient in this family.
V.10 is a 31 year old female, born after an uneventful pregnancy and delivery. In the neonatal period she suffered from recurrent periods of vomiting, for which no definite cause could be found. In infancy she had recurrent bladder infections and upper airway infections. Furthermore, she repeatedly had convulsions without fever. Developmental delay became apparent in early childhood. At the age of 3 years 3 months bilateral inguinal herniae were surgically corrected. She had extreme hyperkinesia and aggressive behaviour, necessitating institutionalisation from the age of 3 years. At present she is severely mentally retarded and lives in an institution for the mentally handicapped.
The results of physical examination at the age of 9 years are summarised in table 1. Many symptoms, such as mental retardation, stocky build, epilepsy, facial dysmorphism (fig 2A, B), and brachymetaphalangism (figs 3 and 4A), fit the Albright hereditary osteodystrophy-like phenotype.
Summary of clinical features

Clinical photographs. (A) Face of patient V.10, aged 2 years. Note round face, prominent forehead, strabismus, deeply set eyes, narrow palpebral fissures, prominent eyebrows (medial), bulbous nose, and thin upper vermilion border. (B) Face of patient V.10, aged 9 years. Note change of features compared to aged 2 years: fine hair, relatively large chin, fleshy earlobes, and somewhat aged appearance. (C) Face of patient V.3, aged 16 years. Note round face, narrow palpebral fissures, bulbous nose, thin upper vermilion border, and macrognathia. (D) Face of patient V.7, aged 15 years. Note full cheeks, somewhat asymmetrical face, deeply set eyes, narrow palpebral fissures, prominent eyebrows, bulbous nose, prominent columella, macrognathia, and prominent ears with thick helices. (E) Face of patient V.19. Note round face, bitemporal narrowing, facial eczematous lesions, sparse eyebrows, narrow and somewhat upward slanting palpebral fissures, deeply set eyes, short, bulbous nose with prominent columella, long philtrum, small mouth, thin upper vermilion border, and macrognathia. (F) Upper part of the body of patient IV.17. Note round, asymmetrical face with generalised alopecia, absent eyebrows, deeply set eyes, narrow palpebral fissures, short, bulbous nose, prominent columella, long philtrum, macrognathia, large and prominent ears with fleshy earlobes, and kyphoscoliosis. (G) Face of patient V.20 (taken from a video film). Note deeply set eyes, narrow and upward slanting palpebral fissures, a short, bulbous nose, long philtrum, thin upper vermilion border, macrognathia, and large ears with fleshy earlobes. (H) Face of patient V.21. Note round face, deeply set eyes, narrow palpebral fissures, prominent lateral eyebrows, a short, bulbous nose with colobomatous alae and a prominent columella, long philtrum, thin upper vermilion border, and macrognathia.

Clinical photographs showing brachymetaphalangism. (A) Left hand of patient V.19, showing shortening of the fourth metacarpal. (B) Hands of patient V.21, showing bilateral shortening of fourth metacarpals.
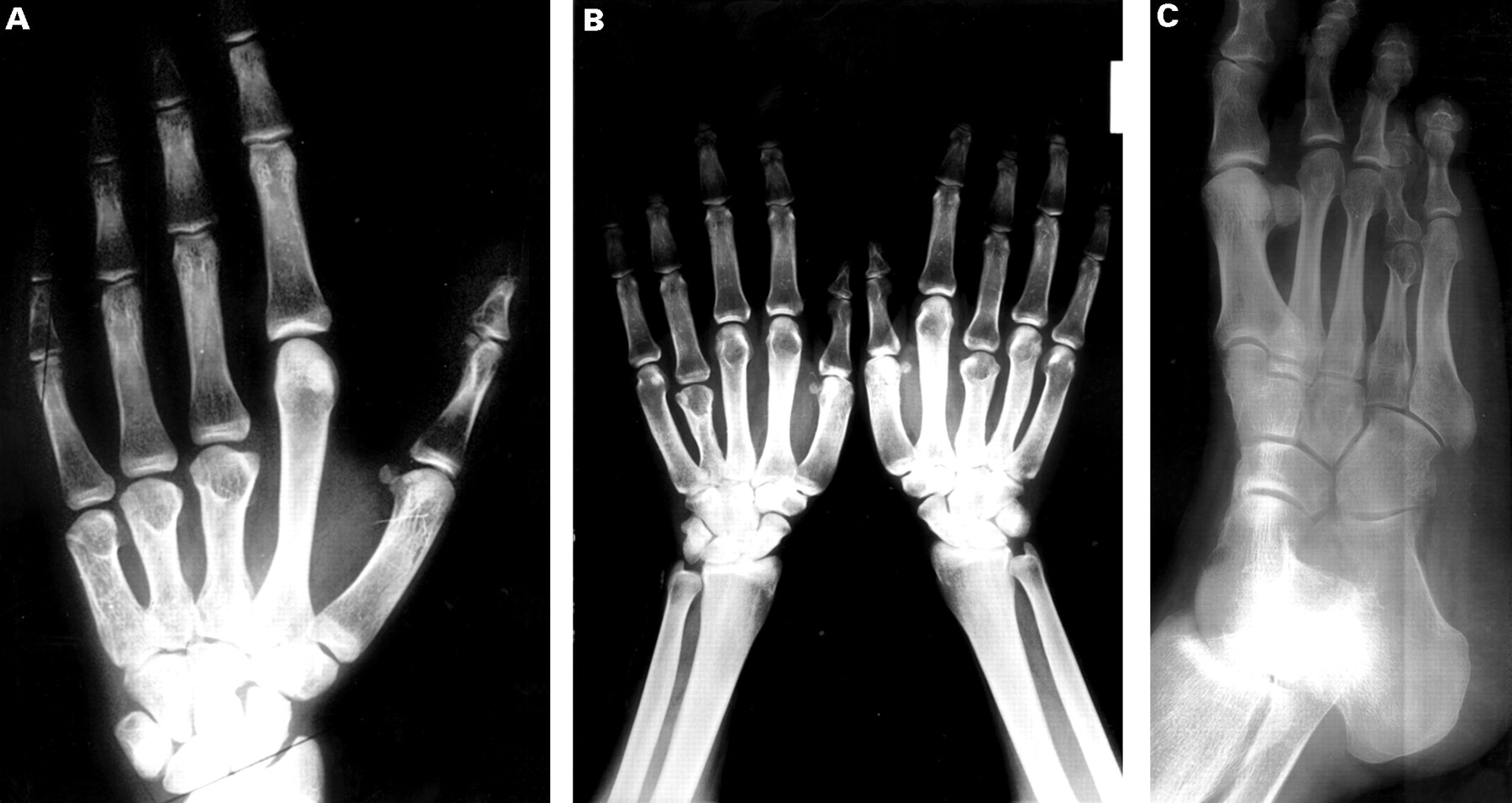
Radiographs. (A) X ray of the left hand of patient V.10 showing extreme shortening of the third to fifth metacarpals. (B) X ray of the hands of patient V.19 showing shortening of left fourth metacarpal and right third and to a lesser extent the fourth metacarpal. (C) X ray of the right foot of patient IV.6 showing shortening of fourth metatarsal.
Internal screen, EEG, 24 hour urinary screen for metabolic disorders, blood investigation (including calcium, phosphate, PTH, urea, creatinine, vitamin D), response to exogenous PTH, and cytogenetic analysis (850 bands) were all normal. X ray showed an advanced bone age (2½ years at the age of 2 years, 10½ years at the age of 9½ years). As with other patients in this family, she did not have any (sub)cutaneous calcifications.
OTHER PATIENTS (V.3, V.7, V.19, IV.6, IV.17, V.20, V.21)
Patients V.3, V.19, and V.21 were alive at the time of this study. Patients IV.6, IV.17, V.7, and V.20 have died, but were carefully examined in the past. All showed symptoms compatible with the AHO-like phenotype. A summary of the major clinical findings is provided in table 1. Clinical photographs of the patients are shown in figs 2C-H and 3A (patient V.19) and B (patient V.21); characteristic radiographs are shown in fig 4B (patient V.19) and C (patient IV.6).
POSSIBLE PATIENTS (III.6, III.19, IV.3, IV.4, IV.19, IV.20, IV.27)
According to anamnestic data, these family members may have been patients, but firm data are lacking. All were reported to have had developmental delay or mental retardation, combined with poor speech in III.19. Apart from IV.3 and IV.4, all were known to have suffered from convulsions.
Skeletal abnormalities are reported less frequently: III.19 was said to have a “disabled left foot and right arm” and IV.27 had short stature (about 1.5 m as an adult).
BEHAVIOURAL PHENOTYPE
Most patients are friendly (V.3, V.19, V.20, V.21), though with advancing age occasional outbursts of aggression (V.19, V.20) and obsessive behaviour (V.20) have been reported. Hyperkinetic behaviour is reported frequently (IV.3, V.7, V.20), sometimes only in childhood (V.19). Patient V.21 self-mutilates when she is frustrated (hand biting).
Patient IV.27 showed destructive behaviour, which necessitated institutionalisation from the age of 7, but patient V.19 lives with her parents and is able to join her family in outdoor activities, such as cycling, swimming, and skating. Patient IV.6 was known to have mental retardation in combination with a psychiatric disorder, probably psychosis.
GENERAL HEALTH
In the neonatal period patient V.19 suffered from unexplained cyanotic episodes, and she had feeding difficulties and a weak cry. During infancy some patients suffered from recurrent upper airway (V.3 and V.20) and ear infections (V.3). Menarche occurred around the age of 14 years (V.19, V.20, V.21).
CAUSES OF DEATH
Most of the (possible) patients died from infectious diseases. Patients III.6, IV.4, and IV.17 died from pneumonia at the ages of 15 years, 16 months, and 40 years respectively, III.19 died from an influenza infection aged 21 years, IV.3 died at the age of 30 years from peritonitis, and IV.20 died from diphtheria at the age of 3 years. Patient V.20 had recurrent pneumonia, but died from choking at the age of 38 years. Patients IV.19 and IV.27 died from status epilepticus, aged 16 and 19, respectively. V.7 died at the age of 19 years from a mediastinitis resulting from a perforated gastric ulcer.
ADDITIONAL INVESTIGATIONS
A CT scan of the brain of V.3 at the age of 19 years showed minimal peripheral atrophy, but was otherwise normal. Plasma calcium and phosphate levels have been found to be normal in IV.17, V.3, and V.7. Investigations in V.3 showed normal levels of urea, creatinine, vitamin D, and intact PTH. At the age of 4 years the bone age in patient V.3 was three years advanced.
Material and methods
GENETIC MARKER STUDIES
Genomic DNA from the index patient (V.10) and her parents was prepared from peripheral blood lymphocytes by standard methods. Thirteen polymorphic 2q markers were typed as described previously.4
CYTOGENETIC STUDIES
Prometaphase chromosomes from peripheral lymphocytes were obtained using a double synchronisation technique.5 Fluorescence in situ hybridisation (FISH) was performed as described previously.6 Slides were examined under a Zeiss Axioplan epifluorescence microscope. For digital imaging microscopy the Cytovision Probe system (Applied Imaging) was used.
Cosmids cCI2-3 (2q36-37.1) and cCI2-60 (2q37.1-37.3),7 and cos2003 were used for in situ hybridisation. Cos2003 is a subclone from half-YAC yRM2003 (D2S446, D2Z4) and recognises subtelomeric DNA in the long arm of chromosome 2 (2q37) and sequences on chromosome 8p23.8
Results
ANALYSIS OF MICROSATELLITE MARKERS
A total of 13 polymorphic markers mapping within 2q37 were analysed, indicating in V.10 a paternal deletion of markers D2S395, D2S90, D2S140, and D2S125 (provisional centromeric to telomeric order according to Wilson et al 4). No informative marker centromeric to D2S395 or telomeric to D2S125 has yet been found. Given this result and the pedigree structure, the possibility of a cryptic translocation involving 2q37 was raised.
FISH STUDIES
In earlier studies, chromosome analysis (both routine and prometaphase) had never shown any chromosomal abnormality in patients IV.6, IV.17, V.3, V.7, V.10 or their parents (fig 5A). Blood lymphocytes from patients V.3, V.10, V.19, and V.21 and their parents IV.1, IV.18, IV.26, and IV.28 were studied with FISH probes. Fibroblasts from V.7 had been stored for over 20 years and were used for FISH analysis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Results of chromosome analysis. (A) Partial karyotype of a patient with a proven deletion of chromosome 2qter, showing chromosomes 2 and 8. On classical cytogenetic investigation there is no difference between the normal and the derivative chromosomes. (B) FISH analysis in a patient; hybridisation of cos2003 to chromosome 2qter and 8pter. One of the chromosomes 2 (arrow) shows a deletion of the telomeric region of chromosome 2q. (C) The same probe hybridised to metaphase chromosomes of a carrier.
In all patients hybridisation of metaphases with the 2q probe (cos2003) showed a signal on one chromosome 2 and, because this probe cross hybridised with chromosome 8p, showed signals on the short arms of both chromosomes 8 (fig 5B). This hybridisation pattern with cos2003 confirmed the molecularly detected deletion of chromosome 2q. Hybridisation with cosmids cCI2-3 and cCI2-60 showed normal hybridisation patterns, indicating a position proximal to the breakpoint on chromosome 2q.
To establish the other chromosome in a presumed translocation, we hybridised cos2003 with metaphases of an obligate carrier (fig 5C). The translocated fragment was located on 8q, indicating the presence of a cryptic translocation t(2;8)(q37.3;q24.3).
In this family, all available patients with Albright hereditary osteodystrophy-like syndrome and mental retardation (V.3, V.7, V.10, V.19, and V.21) showed a terminal deletion of chromosome 2q. No family member showed a terminal deletion of chromosome 8q24.3.
Discussion
A familial cryptic translocation between the long arms of chromosomes 2 and 8, t(2;8)(q37.3;q24.3), is reported, giving rise to a proven subtelomeric deletion of chromosome 2q in five family members and a probable deletion in another 10 members. An AHO-like phenotype is linked to this unbalanced karyotype. The other unbalanced karyotype (trisomy of the subtelomeric region of chromosome 2q and a deletion of the subtelomeric region of chromosome 8q) was not detected. There may be several explanations for its absence. It is possible that one or more of the patients who had died carried this genotype. However, as most members were reported to have had mental retardation and convulsions and some had short stature and brachydactyly, the 2qter deletion is more likely in them. Not all members were examined personally, however, hence some of them may still have had an 8qter deletion/2qter duplication. Alternatively, the 8qter deletion genotype may have a very mild phenotypic effect, if any, and may go unrecognised. This possibility cannot be excluded, as too few family members with a normal phenotype have been karyotyped. A third explanation may be either early fetal loss, which is not known to have occurred repeatedly in the family, but may have gone unrecognised if occurring in the first weeks of pregnancy, or prenatal and perinatal death, known to have occurred nine times in three branches of the family.
The clinical features of AHO are short, stocky build, round face, cutaneous ossification, metacarpophalangeal abnormalities, seizures, hypocalcaemia, and hyperphosphataemia. In the AHO-like phenotype, cutaneous ossification has not been described4 and calcium and phosphate levels are normal. To date there are several case reports describing patients with deletions of chromosome 2q (recently reviewed by Power et al 9). These reports and the description of our family led to the delineation of a syndrome consisting of mental retardation, short stature, brachymetaphalangism, epilepsy, and facial dysmorphism (table 1). Life span appears to be reduced; in several of the present cases death was caused by lower airway infections. Though the patients are generally friendly, their behaviour can be complicated by hyperkinesia, aggression, self-mutilation, or frank psychiatric problems.
It is unknown how the AHO-like phenotype in this family is affected by the duplication of the terminal region of chromosome 8q. To the best of our knowledge, duplications of band 8q24.3 only have never been described. Therefore we are unable to compare a 8qter duplication phenotype, if any, with the summarised phenotype in this family.
The chromosome region for one or more genes for the AHO-like phenotype is defined by microsatellite marker analysis in a series of five patients by Wilson et al.4 The centromeric boundary of the common deleted region is marked by D2S338 and the minimal region of deletion overlap involves D2S125, the most telomeric 2q37 marker used. Microsatellite marker analysis in our family has not helped to define the region, since all markers centromeric to D2S395 were uninformative and the extension of the deletion at the molecular level is therefore unknown. Both testing of additional 2q markers and identification of other AHO-like patients with 2q37 microdeletions may help to narrow down the candidate gene region.
After years of investigation in this family, a cryptic chromosome translocation was found to be the cause of the familial mental retardation syndrome. In patients where an unbalanced karyotype is highly likely, but cannot be detected by routine cytogenetic investigations, we suggest careful comparison with similar cases that have been published or that are in a cytogenetic database. Corresponding cases or specific symptoms entered in a database query may provide a clue regarding chromosome regions that might be involved. Based on the localisation of these regions, probes for FISH analysis can be selected and tested for imbalances. It can be expected that the development of new techniques (for example, 24 colour FISH) will simplify this strategy in the near future, allowing a diagnosis in more of our “unknowns”.
Acknowledgments
We wish to thank Dr Harold C Riethman for the yRM2003 subclone, the Japanese Cancer Research Resources Bank for the cCI2-cosmids, and the family for their willingness to participate in this study.