Article Text

Abstract
The investigation of mtDNA disease can be relatively straightforward if a person has a recognisable phenotype and if it is possible to identify a known pathogenic mtDNA mutation. The difficulties arise when no known mtDNA defect can be found, or when the clinical abnormalities are complex and not easily matched to those of the more common mitochondrial disorders. We will describe here the difficulties that can be encountered during the identification of pathogenic mtDNA mutations and the approaches that can be used to confirm, or eliminate, a likely pathogenic role, in either single gene diseases or in multifactorial disorders.
- mitochondrial DNA
- phylogenetic analysis
- Leber’s hereditary optic neuropathy
- Alzheimer’s disease
Statistics from Altmetric.com
The accumulation of mitochondrial DNA point mutations and the mitochondrial genetic “clock”
Because mitochondrial DNA accumulates mutations much more rapidly than nuclear DNA, the mtDNA sequence of any one person from the world wide population differs from that in another person by an average total of 25 base pair substitutions (Andrews et al, unpublished observations). This degree of polymorphism is useful in forensic medicine,1 2 in the construction of mtDNA phylogenies,3 and in the analysis of population migrations.4-7
There have been numerous attempts to measure the rate of evolution of the human mitochondrial genome. The most common approach is to construct a phylogenetic tree from a collection of mtDNA sequences, use the tree to estimate the average number of sequence changes/genome (branch length) that have occurred since the time of the last common maternal ancestor, and then to calculate the rate of evolution relative to some “benchmark”, such as the time of the human-chimpanzee divergence. This phylogenetic approach is dependent upon several factors, including an accurate value for the benchmark time and a realistic model of sequence evolution that is used for phylogenetic tree construction, both of which are highly prone to uncertainty and inaccuracy.8 9 For example, different regions of the mitochondrial genome evolve more rapidly than others. Thus, the hypervariable sequences in the non-coding displacement loop evolve much more rapidly than the coding regions.10 This site variability has profound effects on phylogenetic estimates of divergence rates.11 It is not surprising, therefore, that different laboratories report different rates of mtDNA evolution.2 10 12 Other processes may also contribute to this problem, including selection, positive or negative, operating on some sequence changes, non-independence of mtDNA sequence changes, and the possibility of a temporally episodic clock (that is, evolution occurring as a result of bursts of mutations13).
Unfortunately, this high degree of polymorphic variability creates major problems when trying to ascribe pathogenicity to a new base change, because the pathogenic mutation is typically “buried” within a background of multiple sequence changes. In addition, polymorphisms may be relatively rare themselves and cosegregate with disease, confounding identification of the pathogenic mutation. When is a mtDNA sequence change pathogenic and when is it simply a benign polymorphism?
Although the publication of the human mtDNA Cambridge Reference Sequence (CRS) in 1981 set the “gold standard” for sequence comparisons,14 it is important to recognise exactly what this sequence represents. Firstly, it is primarily the human mtDNA, but the bovine mtDNA sequence was used at some ambiguous sites although the latter are not completely specified. Secondly, the human sequence was obtained primarily from a single person of European descent who has subsequently been found to have a rather unusual mitochondrial genome. In addition, the HeLa mtDNA sequence, which is known to be African-American in terms of ethnic origin, was used for some regions, again unspecified. Perhaps not surprisingly, therefore, there have been a number of “errors” in the CRS which have emerged over the last decade.
Because of the unusual features of the CRS and because of the high frequency of benign mtDNA polymorphisms, it is extremely important to compare previously unidentified, putative pathogenic mutations to a number of appropriate reference sequences, before reaching any conclusions about pathogenicity. The strongest proof of pathogenicity comes when the mutation has been shown to arise numerous different times on different haplotype “backgrounds” within the human population. In addition to the information in online mtDNA sequence databases, we have determined the complete sequence for more than 50 mitochondrial genomes from subjects of European descent (Andrewset al, manuscript in preparation).
When is a mtDNA mutation significant?
One criterion for pathogenicity is heteroplasmy. If a mtDNA rearrangement or point mutation is heteroplasmic, then this condition suggests either that it arose relatively recently (because it has not yet had time to become homoplasmic), or that humans cannot tolerate the mutation in the homoplasmic state.15 A more compelling criterion comes from measurement of the percentage level of mutant mtDNA in clinically affected tissues, and comparison of this level to that in unaffected tissues. If the mutation load is higher in the former, then this is evidence for pathogenicity. This approach has been refined even further. Patients with pathogenic mtDNA defects often have a mosaic pattern of cytochrome c oxidase deficiency on muscle histochemistry. Through the isolation of individual skeletal muscle fibres, it has been possible to show that the percentage level of a novel base change is significantly higher in cytochrome c oxidase deficient fibres, when compared to normal fibres.16-18 Because many novel base changes that cause neurological disease are heteroplasmic, this approach has proved to be very powerful.
Unfortunately, as a number of mtDNA point mutations are homoplasmic, it can be extremely difficult to prove that a novel base change causes disease. Under these circumstances, the application of nuclear genetic criteria may be helpful. Regions of DNA sequence that are similar in many species are assumed to be functionally important (evolutionarily conserved sites). As a result, base changes that have not been tolerated during evolution are more likely to be pathogenic, particularly if they result in an amino acid substitution that could plausibly deleteriously affect the structure or function of the gene product. This approach only holds true, with any degree of certainty, if the mutation is only found in clinically affected subjects and not in unaffected controls. This caution leads one back to the question: what is a good control? As discussed in the previous section, one should use a number of reference mtDNA sequences, including as many as possible from normal controls that are closely related, in the phylogenetic sense, to the mtDNA that carries the putative pathogenic mutation.
Another complexity of mitochondrial disorders and their genetic basis is that a subject’s mitochondrial genotype is not an unchanging entity throughout the life span. The mitochondrial genome acquires somatic mutations during the normal life span. Despite the importance of each mitochondrial gene, mtDNA is not associated with protective histones, mitochondria have limited DNA repair mechanisms, and the mitochondrial respiratory chain is a potent source of DNA damaging free radicals.19 MtDNA mutations accumulate in postmitotic tissues such as brain20 and skeletal muscle.21 The rate of accumulation may be much faster in certain disease states (such as Alzheimer’s disease in brain,22 myocardial ischaemia,23 and inflammatory muscle disease24 25). The mean level of these mutations in individual tissues is low (<1%). However, single cell studies have shown that the mutations may clonally accumulate to high levels in ageing tissues leading to mitochondrial dysfunction,26 but the importance of these so-called “secondary” mutations, and their role in ageing and neurodegenerative disease, remains to be determined.
MtDNA mutations do not exert their effects in isolation
Up to this point, we have been concerned with specific mtDNA mutations that directly cause specific diseases, that is, the mtDNA mutation is both necessary and sufficient for manifestation of the clinical abnormalities. However, in many cases, the situation is more complex than this and secondary aetiological factors, genetic or environmental, are involved (fig 1). For example, in one study the 1555 point mutation in 12S mitochondrial ribosomal RNA (rRNA) was responsible for up to 27% of cases of non-syndromic sensorineural deafness.27 The mutation alters the aminoglycoside binding site of the 12S rRNA, rendering affected subjects susceptible to environmental effects of ototoxic aminoglycosides. The aetiology of Leber’s hereditary optic neuropathy (LHON) is more complex. Over 95% of cases of LHON are the result of one of three point mutations that affect mitochondrial complex I genes at nucleotide positions 11 778, 14 484, and 3460 of the mtDNA L strand.28 These mutations have arisen multiple times in the human population,29which firmly establishes their primary pathogenic role. However, the penetrance is incomplete and it is still not clear why only 50% of males and 10% of females develop visual loss.30 Attempts to identify a relevant locus on the X chromosome have not been successful,31 and the difference may be the result of gender related anatomical and physiological differences.32The incomplete penetrance provides strong evidence that there must be additional factors, genetic or environmental or both, which augment or modulate the pathogenic phenotypes of the primary LHON mutations. For example, it is now recognised that the toxic effects of alcohol and tobacco increase the risk of visual failure in those who inherit LHON mutations.33 The possible secondary genetic interactions, however, are even more complex and less firmly established.
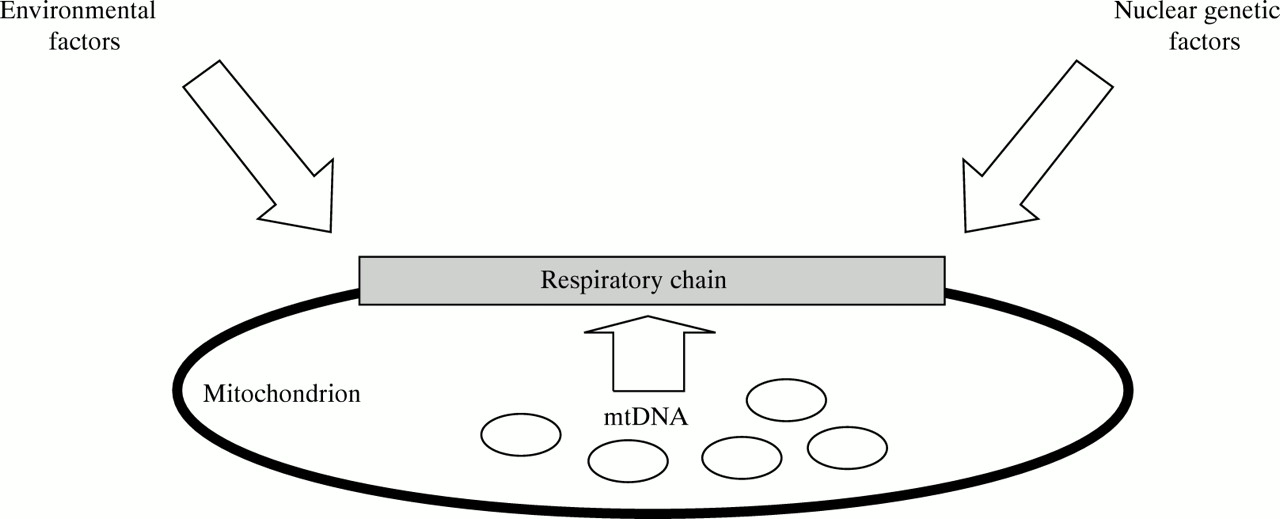
Mitochondrial DNA defects do not exert their effects in isolation. Nuclear genetic and environmental factors influence the expression of mtDNA and respiratory chain function.
In 1991, Johns and Berman34 noted that two nucleotide substitutions (4216 and 13 708) were more common in patients with the 11 778 LHON mutation than in normal controls. Further studies showed that the 4216 and 13 708 substitutions were also more frequent in patients with the 14 484 LHON mutation than in controls,35 but there was no such association with the 3460 LHON mutation.36 As a result, Johns and Berman34 suggested that the 4216 and 13 708 substitutions were “secondary” LHON mutations. One difference between the primary and “secondary” mutations is that they alter less stringently conserved amino acid residues, and they are found in between 10 and 15% of all Europeans,37 and they do not cause LHON on their own. The key question remains whether these “secondary” substitutions affect the expression of LHON.
One systematic way of comparing different mtDNA sequences is through phylogenetic analysis.38 This is one way of deducing the maternal family structure and history of a contemporary population, because it provides information on the number of sequence changes that have occurred and their relative temporal order. This approach is based upon the assumption that similar sequences share a recent common maternal ancestor. By contrast, the common ancestor for two divergent sequences must have occurred at a much earlier time. Software (for example, PHYLIP is available from J Felsenstein at the University of Washington (Seattle, WA); MEGA is available for a small fee from Professor M Nei at Pennsylvania State University (University Park, PA); and PAUP* is an expanded and updated version of PAUP that is available commercially from Sinauer Associates (Sunderland, Massachusetts)) will construct a phylogenetic tree based on the observations in the current population. The tree that requires the fewest sequence changes (the most parsimonious) is assumed to be the most likely representation of the maternal ancestry of the population under study. When this approach was applied to a large number of LHON pedigrees, the most parsimonious phylogenetic tree indicated that the 4216 and 13 708 mutations arose once, and that they defined a monophyletic cluster (fig 2). However, the primary pathogenic LHON mutations arose multiple times within this cluster. What is the mechanism behind this association? The phylogenetic analysis effectively rules out the possibility that the 11 778 and 14 484 mutations predispose to multiple origins of the 4216 and 13 708 mutations. The fact that the 11 778 and 14 484 mutations apparently arose multiple times excludes a major founder effect as the basis of the association. Two other possibilities remain. The 4216 and 13 708 substitutions may alter the penetrance of the primary LHON mutations, increasing the clinical presentation.37 Alternatively, these secondary LHON mutations may predispose subjects to the origin and fixation of the 11 778 and 14 484 mutations. The details of this discussion are not as important here as the principle that they illustrate. LHON has a well defined, relatively simple phenotype. The aetiological complexity is manifold when we consider other more common multifactorial disorders, such as the neurodegenerative diseases.

{kind=link}
{kind=link}
The secondary LHON mutations arise within a single phylogenetic cluster. A simplified phylogenetic tree showing the early origin of the 4216 mutation which is present within all branches of the cluster. In one branch the 4917 mutation arose and in another branch the 13 708 mutation arose. The 13 708 branch is further divided by the 15 257 mutation which is relatively recent in origin.
MtDNA haplotypes and multifactorial diseases
Epidemiological studies indicate that the risk of developing Alzheimer’s disease (AD) and Parkinson’s disease (PD) is greater in the offspring of mothers who develop the disease than among the offspring of fathers who develop the disease.39-41Because mtDNA is inherited strictly down the maternal line, a mtDNA abnormality could be one explanation for these observations. However, it is important to recognise that this “maternal bias” could arise for a number of other reasons. For the late onset disorders, it may simply reflect the relative longevity of females. Alternatively, pseudomaternal transmission may be the result of anatomical or physiological sex differences, or an X linked locus leading to an increased prevalence of females with the disease, although there is convincing evidence that males and females are affected at equal frequencies. The possibility that females also may be more likely to present to medical attention may influence the apparent inheritance pattern. Finally, methylation of autosomes may create the impression of sex linked inheritance (genomic imprinting). It must be stressed that, so far, no satisfactory explanation for these maternal effects has been forthcoming.
The most obvious explanation for the maternal effect in the inheritance of AD and PD is that a mtDNA mutation(s) plays an aetiological or pathogenic role in these disorders. One avenue of investigation has obtained evidence for specific respiratory chain defects in AD and PD patients,42-46 and that these defects can be transferred to cybrid cells.47 48 These experiments, and their interpretation, have been controversial.49 In an alternative approach, there have been several studies that have screened for mtDNA mutations in patients with AD and PD, but the results have not been consistent. For example, three population screening studies have found that a substitution at nucleotide 4336 in the mitochondrial tRNA glycine gene was more prevalent in AD and PD patients when compared to controls.50-52. However, other studies have not found a significant association.53 Even in the former studies, this putative mtDNA mutation accounts for no more than 5% of the AD or PD cases. These findings raise the possibility that mtDNA sequence variants may interact with nuclear and environmental factors, leading to an increased risk of developing neurodegenerative disease.50 It is also possible that the cumulative effect of a number of sequence variants may compromise mitochondrial function, although it not clear why the respiratory chain defects lead to AD and PD, rather than the more typical abnormalities that are found in mitochondrial disorders. Alternatively, the mtDNA haplotype may lead to the formation of secondary mitochondrial mutations.32 However, these investigations must be interpreted carefully. Nuclear pseudogenes may be a potential source of errors when looking for potential pathogenic mtDNA mutations. This possibility was illustrated by the recent report of a higher frequency of heteroplasmic mutations in cytochrome coxidase genes in AD,54 which are now known to be pseudogenes.55 56
The central question is, do these statistical associations represent a genuine increased risk? To continue with the 4336 mutation as an example, this base substitution is uncommon in the general population (estimated at <1%51) and a large disease group may be necessary to show a clear difference between AD patients and controls. The most difficult aspect of a study of this type is the selection of an appropriate control group. For neurodegenerative diseases, this means age and sex matched subjects with no histopathological evidence of the disease in question. However, even these efforts cannot be perfect because of the late onset and the presence of insidious, subclinical disease. That is, there will always be a substantial number of disease free controls who will subsequently develop AD or PD. If the controls are inadequate, then any disease associated effect can be diluted down to statistical insignificance or artificially inflated to statistical significance. In any case, the association of a particular mtDNA sequence variant with a particular disease is not an inviolate indicator of aetiological significance. The mtDNA sequence may act as a surrogate marker for a nuclear genetic defect, particularly for isolated or inbred populations that have experienced a marked founder effect.57 58 In such populations, there should be a statistically significant association between mitochondrial and nuclear genotypes for several generations. This scenario is one likely explanation for the association between the nt 16 519 D loop polymorphism and hypertriglyceridaemia in the Oji-Cree people of North America.59 Similarly, a particular mtDNA haplotype may signal, through a founder effect, a population subgroup that has inherited a group of detrimental (or protective) nuclear genes. This effect is one explanation for the apparent relative longevity of one subgroup of the Japanese population.60 61 Although the mtDNA sequence variants may not be directly related to the traits that they accompany, the association may be genuine and it may help in diagnosis and targeting new therapies.
Have we ignored the nucleus?
As we come to understand more about the mitochondrial genome and its expression, there is an accumulating body of evidence that supports a role for nuclear genes in the pathogenesis of a number of mitochondrial diseases. The phenotypic consequences of the A3243G point mutation, on the whole, tend to be consistent within individual matrilineal pedigrees. Thus, one family may suffer predominantly from diabetes and deafness,62 a second from cardiomyopathy,63 a third from CPEO,64 and a fourth from encephalomyopathy. It is difficult to explain this trend solely on the basis of tissue specific segregation of heteroplasmic mtDNA mutations, and nuclear genetic influences are an attractive candidate for further experimental and theoretical analysis. The importance of these clinical observations has been augmented by the cybrid cell work, where the nuclear background influences whether the percentage level of mutant mtDNA drifts up or down in culture. It may be useful, therefore, to think of mtDNA mutations as a “mismatch” between the nuclear and mitochondrial genomes.65 For some mutations (such as the A3243G point mutation), the mtDNA mutation in itself is pathogenic, but different nuclear genetic backgrounds may influence the nature and severity of the disease. This suggested mechanism of pathophysiology may provide a target for the design of novel therapies.
Conclusions
MtDNA mutations are an important cause of human genetic disease. Although these mutations cause a daunting spectrum of diseases, simply having a high index of suspicion in patients with multisystem disorders often aids diagnosis. The investigation of these patients may be complex, but provided that it is approached in a rigorous and systematic way, the yield of positive diagnoses is often high. It is important to identify these patients because of the possibility of preventing complications, and because of the distinct genetic and prognostic counselling which can be given. Confident identification of the novel disorders can be difficult, and the unwary are often led astray. Even for diseases with an obvious maternal inheritance pattern or identified respiratory chain defect, the pathogenic role of mtDNA sequence variants may be difficult to establish, and we urge caution when pointing the finger at any part of the mitochondrial genome. These complexities and uncertainties notwithstanding, the past 10 years have seen a remarkable advance in the genetic analysis of mitochondrial diseases.
Acknowledgments
PFC is a Wellcome Trust Research Fellow. NH acknowledges support from the National Eye Institute (RO1 EY10758), the John Sealy Memorial Endowment Fund, and The Wellcome Trust. RA is a MRC Research Training Fellow. DMT is supported by The Wellcome Trust and the Muscular Dystrophy Group of Great Britain.