Article Text

Abstract
Silver-Russell syndrome (SRS) shares common features of intrauterine growth retardation (IUGR) and a number of dysmorphic features including lateral asymmetry in about 50% of subjects. Its genetic aetiology is complex and most probably heterogeneous. Approximately 7% of patients with SRS have been found to have maternal uniparental disomy of chromosome 7 (mUPD7). Genomic DNA samples from five SRS patients with mUPD7 have been analysed for common regions of isodisomy using 40 polymorphic markers distributed along the length of chromosome 7. No regions of common isodisomy were found among the five patients. It is most likely that imprinted gene(s) rather than recessive mutations cause the common phenotype. Heterodisomy of markers around the centromere indicated that the underlying cause of the mUPD7 is a maternal meiosis I non-disjunction error in these five subjects.
- Silver-Russell syndrome
- mUPD
- heterodisomy
- isodisomy
Statistics from Altmetric.com
Silver-Russell syndrome (SRS) was first described in 1953-54 and is a combination of intrauterine and postnatal growth retardation, relative sparing of cranial growth, triangular face, downturned corners of the mouth and, in a large proportion, clinodactyly V and facial, limb, or truncal asymmetry.1 The postnatal growth pattern is characteristic with persistent short stature, marked leanness, and a tendency for early, or at least prompt, puberty with further restriction of adult height.2 3
The genetic aetiology is complex with chromosomes 8, 15, 17, and 18 plus autosomal recessive, dominant, and sex linked modes of inheritance being implicated at various times. However, there have now been three major studies of maternal uniparental disomy 7 (mUPD7) associated with SRS.4-6 In total, nine cases of mUPD7 have been reported out of 99 patients studied. Of these nine, three were reported as showing complete isodisomy, two complete heterodisomy, and four a mixture of hetero- and isodisomy. However, there have also been studies where no cases of mUPD7 were identified,7-9 and the true prevalence of mUPD7 is probably around 7%.
In addition there have been five single case reports of mUPD7 associated with pre- and postnatal growth retardation.4 10-13 Two of these10 11presented with cystic fibrosis where only the mother was a carrier of a CFTR mutation and the probands were homozygous for the same CFTR mutation because of maternal isodisomy of chromosome 7; one of these patients had some dysmorphic features compatible with SRS. A third12 had a similar situation affecting COL1A2; the remaining two were growth retarded without extra features. Out of these five, three were reported as isodisomic,4 10 11 one heterodisomic,13 and one with mixed hetero- and isodisomy.12
Uniparental disomy can cause a phenotype by one of two mechanisms or a combination of both. If the mUPD is isodisomy then the duplication of one grandparental chromosome may expose any recessive gene(s) carried at a particular locus, as in the earlier case reports of exposed recessive mutations in CFTR10 11 or COL1A2.12 If the mUPD is heterodisomy, with both grandparental chromosomes present, this mechanism cannot apply and the phenotype is more likely to be the result of a disturbance of the expression of imprinted genes. A gene expressed only from the paternal allele carried on chromosome 7 would not be expressed in a subject with mUPD7 as the gene is effectively deleted. In contrast a maternally expressed gene would be expressed from both alleles, instead of the usual one, and could have phenotypic effects as a result of an excess of gene product.
One approach to the study of the aetiology of SRS is to analyse chromosome 7 for imprinted regions or genes and to search for mutations in these genes in SRS cases without mUPD7. To be certain that the imprinted gene approach is valid, common isodisomic regions need to be ruled out, as this could imply a recessive gene effect. To date, published data show heterodisomy at the majority of marker loci. However, the most distal 7p marker (D7S21) is that reported by Preeceet al 5 and in these three cases this marker was isodisomic in two cases and uninformative in the third. This allows the possibility of a small isodisomic region including D7S21 and more distal markers. The present study is an extension to five cases with an increased density of polymorphic markers to clarify the distribution of hetero- and isodisomy.
Materials and methods
Out of our cohort of 53 SRS patients, five have been found with mUPD7; these form the basis of this more detailed study. All five conformed to the clinical and cytogenetic criteria previously described.5 DNA was extracted from blood lymphocytes obtained from probands and parents using conventional methods.14
Two VNTR probes (D7S21 and D7S22, Cellmark Diagnostics, UK) and 38 polymorphic di- and tetranucleotide microsatellite repeat markers (HGMPRC, Cambridge, UK and Research Genetics Inc, AL, USA) were used. Their cytogenetic locations and genetic distances from the p terminus are given in fig 1. For both VNTRs, genomic DNA was restricted withHinfI followed by Southern blotting and hybridisation using standard methods. Microsatellite repeats were analysed either using fluorescently labelled primers and standard semiautomated methods15 or radioactive PCR.16Not every proband was studied for all the markers owing to limited supplies of DNA in one or other family member. Paternity in each case was confirmed using four polymorphic markers from chromosomes 2, 6, 11, and 17.

Schematic diagram to show the distribution of the polymorphic markers used in this study. On the left, an ideogram of chromosome 7 is shown with the cytogenetic localisations of the markers indicated, where known (data from The Genome Database,http://www.hgmp.mrc.ac.uk/gdb/gdbtop.html). Columns 2 and 3 show the genetic distances between markers. As there is no integrated map that localises all 40 markers used, two sources were used. That on the left uses the Genetic Location Database (http://cedar.genetic.soton.ac.uk/pub/chrom7/map.html) and that on the right uses data provided by Research Genetics Inc, which depend on the Comparative Human Linkage Center map (http://www.chlc.org/). The distances from the Genetic Location Database have been used subsequently with the positions of markers D7S1802 and D7S2195 estimated by interpolation using the CHLC map. Maximum heterozygosity is shown in column 4.
The study was approved by the Joint Research Ethics Committee of Great Ormond Street Hospital and the Institute of Child Health, approval number 1278.
Results
PATERNITY
Analysis of VNTRs or microsatellite repeat markers on chromosomes 2, 6, 11, and 17 were all consistent with paternity as stated in all five families.
MARKERS ON CHROMOSOME 7
A total of 40 polymorphic markers distributed along the p and q arms of chromosome 7 were studied. On average this represented a marker every 4.5 cM. Haplotypes for each of these markers were determined in the five probands and their parents so that the parental origins could be determined at each locus. Approximately half of the possible haplotypings were informative for parent of origin and all 102 informative haplotypes supported mUPD7. At no locus was there a detectable paternal allele. The determination of the distribution of iso- or heterodisomy depended on the comparison of proband and maternal genotypes. In this study, 157 were informative as to which maternal chromosomes were transmitted to the proband. A marker is described as informative if it informs the assignment of iso- or heterodisomy and not parent of origin. The distribution of disomy is shown in fig2.

{kind=link}
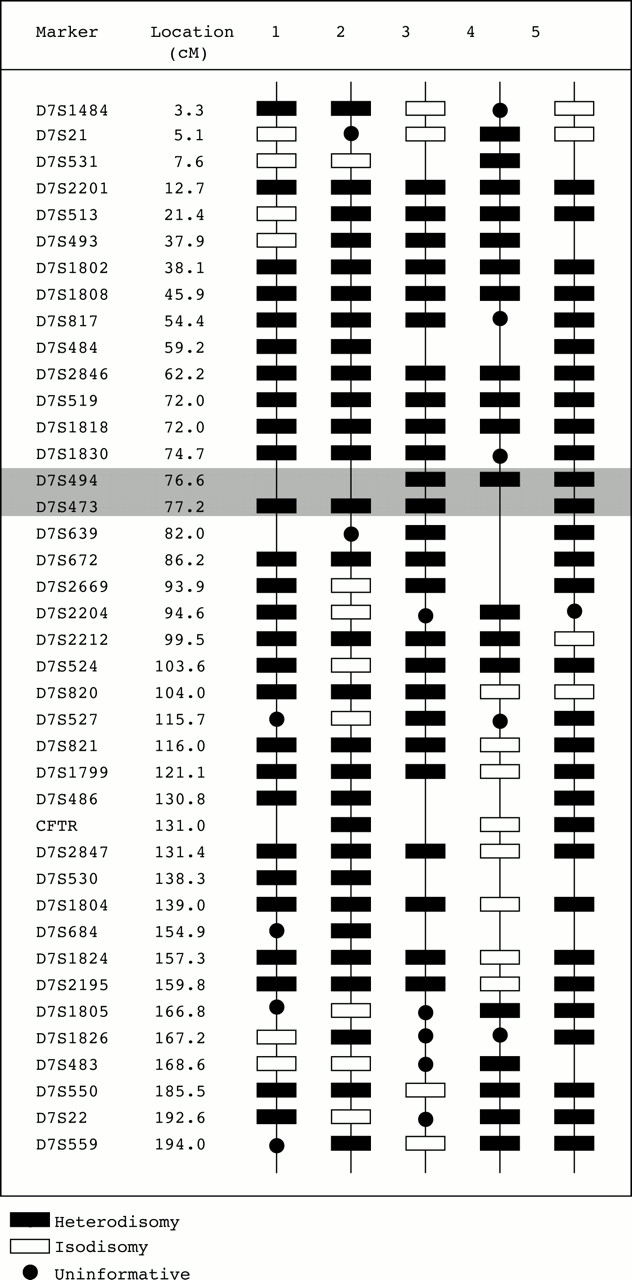{kind=link}
The distribution of hetero- and isodisomy on chromosome 7 for the five probands with SRS and mUPD7. The five columns on the right of the figure show the distribution of iso- and heterodisomy in a schematic manner. Markers that were not studied in a particular subject have no symbol. The second column shows the distances in cM of each marker from pter. The shaded area indicates the approximate location of the centromere.
For most informative loci, both of the maternal chromosomes 7 were present indicating heterodisomy. For all other informative loci only one maternal allele was detected indicating that the same chromosome 7 had been transmitted twice resulting in isodisomy. In each mUPD7 subject, a mixture of both heterodisomy and isodisomy was detected (fig2). The order of markers has been based on a consensus between two sources of genetic mapping data (see legend). Despite the ambiguity of the distances between several of the markers, it is clear that no isodisomic regions are common to the five mUPD7 probands. The most telomeric region of 7p that may have contained a small common isodisomic region (D7S21-7pter)5 shows heterodisomy in proband 4 and also at D7S1484, which is 2 cM more distal, in probands 1 and 2.
CENTROMERIC MARKERS
Two dinucleotide markers have cytogenetic localisations spanning the centromeric region (D7S473 and D7S494) and in all five probands indicated heterodisomy when informative. Two markers on the long arm localised close to the centromere (D7S639 and D7S672) are also consistently heterodisomic when informative. This implies that the non-disjunction event that occurred before trisomic rescue was a meiosis I error.
Discussion
The principal aim of this investigation was to analyse mUPD7 in five subjects for common regions of isodisomy. This would clarify whether exposure of recessive genes or disruption of the expression of imprinted genes, or some combination of the two, underlies the mechanism whereby mUPD7 was associated with the clinical phenotype of SRS. For an exposed recessive gene to be the underlying mechanism there would need to be a consistent region of isodisomy on chromosome 7 in all five probands. From fig 2 it is clear that this does not occur and for every marker there is at least one subject with heterodisomy.
Could there be a common region of isodisomy that has been missed in this investigation? For this to have occurred there would need to be a double recombination between two of the markers studied. To assess the probability of this, it is necessary to examine the genetic distances between markers. This is made more difficult by the fact that there is no completely integrated genetic map that includes all the markers used in the study. Fig 1 shows the two most useful maps with the Genetic Location Database being given priority for initial ordering of the markers, as it included the majority of markers. First using the data from the Genetic Location Database, the greatest genetic distance between markers is that between D7S483 and D7S550 which is 17.0 cM. The probability of a double recombination being present in this interval is therefore 0.172=0.029 for one proband and to occur in the same region in all five probands is 0.0295=2.0 × 10-8. Using the CHLC map, the greatest distance is between D7S513 and D7S1802 which is 20.0 cM, giving a probability of 0.04 for one subject and 1.0 × 10-7 for all five. It is, therefore, clear that such an occult double recombination is extremely unlikely over the region of chromosome 7 bounded by markers D7S1484 and D7S559.
It is also relevant to explore the number of recombination events required to explain the data. In the cases of probands 1, 3, 4, and 5, the number of chiasmata required is between two and five, which is possible for a chromosome of this size. However, in the case of proband 2, a minimum of 14 chiasmata are required to explain the data, which seems unlikely. It is possible that the localisations of one or more markers may be inaccurate. For example, if D7S1805 and D7S1826 were reversed, two fewer recombinations would be required and this would not alter the interpretation in the other four probands; the same applies to D7S524 and D7S820. Alternatively, in the case of one or more of the apparently isodisomic markers there may have been preferential amplification of one allele,17 possibly owing to a polymorphism in the primer site of the unamplified allele. A third, but less likely explanation could be an association of excessive crossover formation with meiotic non-disjunction, but this is less likely as the reverse is usually true.18
A secondary aim of this project was to study the pattern of hetero- and isodisomy around the centromere in order to explore the underlying mechanism of the mUPD7. There were two markers whose localisations span the centromere (D7S473 and D7S494) and a further two flanking the centromere on the long arm (D7S639 and D7S672). In all informative haplotypes these markers were heterodisomic, which indicates that the most likely underlying mechanism is maternal meiosis I non-disjunction leading to trisomy 7,18 with subsequent trisomic rescue.
In summary, the mUPD7 associated with the SRS phenotype is probably the result of a meiosis I non-disjunction followed by trisomic rescue with loss of the paternal chromosome. The expression of the clinical phenotype is still most likely to be the result of the presence of an imprinted gene or genes on chromosome 7. If the causal gene or genes were paternally expressed, then in mUPD7 there is effectively deletion of the gene or genes and the SRS patients without mUPD7 would be explained by imprinting mutations or mutations/deletions of the paternal copy. Alternatively, if the gene were maternally expressed, the mUPD7 patients would have two gene copies capable of expression, leading to an excess of the gene product. The mechanism in the non-UPD patients is more complex requiring either microduplications of the imprinted region of the chromosome of maternal origin or imprinting mutations of the paternal copy that allow expression of this allele.
Acknowledgments
A Dunhill Medical Fund Research Fellowship supports SNAA, ELW is the recipient of an Action Research Training Fellowship, and KAA was supported by a grant from The Child Growth Foundation. We are grateful to Dr Sue Price who visited almost all the families studied and provided clinical data and samples. We are also grateful to Dr Richard Trembath and Lee Clough of the University of Leicester who assisted with the automated dinucleotide repeat marker analysis.