Article Text

Abstract
Bardet-Biedl syndrome (BBS) is an autosomal recessive condition characterised by rod-cone dystrophy, postaxial polydactyly, central obesity, mental retardation, hypogonadism, and renal dysfunction. BBS expression varies both within and between families and diagnosis is often difficult. We sought to define the condition more clearly by studying 109 BBS patients and their families, the largest population surveyed to date. The average age at diagnosis was 9 years, which is late for such a debilitating condition, but the slow development of the clinical features of BBS probably accounts for this. Postaxial polydactyly had been present in 69% of patients at birth, but obesity had only begun to develop at around 2-3 years, and retinal degeneration had not become apparent until a mean age of 8.5 years. Our study identified some novel clinical features, including neurological, speech, and language deficits, behavioural traits, facial dysmorphism, and dental anomalies. In the light of these features we propose a revision of the diagnostic criteria, which may facilitate earlier diagnosis of this disorder. We present evidence for an overlapping phenotype with the Laurence-Moon syndrome and propose a unifying, descriptive label be adopted (polydactyly-obesity-kidney-eye syndrome).
We report an increased prevalence of renal malformations and renal cell carcinoma in the unaffected relatives of BBS patients and suggest that these may be a consequence of heterozygosity for BBS genes. Our findings have important implications for the care of BBS patients and their unaffected relatives.
- Bardet-Biedl syndrome
- diagnosis
- renal malformation
- heterozygotes
Statistics from Altmetric.com
Bardet-Biedl syndrome (BBS) (MIM 209900) is an autosomal recessive condition with a wide spectrum of clinical features. The principal manifestations are rod-cone dystrophy (sometimes called atypical retinitis pigmentosa), postaxial polydactyly, central obesity, mental retardation, hypogonadism, and renal dysfunction.1 2 Other features, not always present, include hepatic fibrosis, diabetes mellitus, reproductive abnormalities, endocrinological disturbances, short stature, developmental delay, and speech deficits. BBS is distinguished from the much rarer Laurence-Moon syndrome, in which retinal pigmentary degeneration, mental retardation, and hypogonadism occur in conjunction with progressive spastic paraparesis and distal muscle weakness, but without polydactyly.3
BBS is genetically heterogeneous, with four loci mapped to date. These are BBS1 (11q13),4 BBS2 (16q22),5 BBS3 (3p13),6 and BBS4 (15q21).7 We have recently shown that the BBS1 locus is involved in ∼45% of affected white families.8 The BBS4 locus appears to be the next most common,9 but there are several families of Middle Eastern and Asian origin which do not show linkage to any known locus. Genotype-phenotype correlations between the various loci do not show obvious differences, with the possible exception of minor effects on growth.8
We had noted certain, previously unreported clinical complications in a few BBS patients and were interested in establishing their prevalence. We undertook a survey of 109 BBS patients resident in the UK and their families, the largest such group reported to date. We present the results of this survey and discuss the findings and their implications for improved diagnosis and management of BBS.
Methods
QUESTIONNAIRE
A seven page questionnaire, requesting details of the patient’s birth and delivery, age at diagnosis, and diagnosing clinician was used. Information was sought regarding height, weight, presence or absence of limb abnormalities (polydactyly or brachydactyly), presence of rod/cone dystrophy (atypical retinitis pigmentosa) or other eye conditions, renal abnormalities, developmental delay, and involvement of other organs. We also enquired whether any other, apparently unaffected, family members exhibited any manifestations associated with BBS. This questionnaire was sent out together with one from the Laurence-Moon-Bardet-Biedl Society of Great Britain (LMBBS), designed to obtain practical information regarding the level of education received and the social interests of patients and their families, together with details of financial support, involvement with support groups, use of special aids or clothing, and careers advice. The information obtained by the LMBBS in this way has been of great assistance to newly diagnosed patients and their families seeking advice and support from the Society.
PATIENTS
Patients were identified either through the LMBBS or through the Guy’s Hospital Bardet-Biedl Register. One hundred and twenty five prepaid reply questionnaires were sent out directly to patients and their relatives and 112 replies (90%) were received. Patients were offered telephone help in completing the form if they had a specific query or if they were unable to enlist the help of a sighted person. In all cases where there were ambiguities or points of interest on the completed form, the patient (or a relevant relative) was contacted directly; 44 cases were visited at home or seen in the Genetics Clinic.
Using the diagnostic criteria proposed by Schachat and Maumenee,10 which require the presence of four out of five principal features (retinal degeneration, mental retardation, obesity, polydactyly, hypogonadism), the BBS status of the respondent (affected versus unaffected) was determined. Only those respondents clearly satisfying these diagnostic criteria (109 of the 112 who replied, or 97%) were included in the analysis.
STATISTICS
The mean, standard deviation, and range were calculated where appropriate. Student’s t test was used for comparison of means and a significance level of 5% (p=0.05) set.
Results
PATIENTS
One hundred and nine BBS patients, 47 females and 62 males, met the diagnostic criteria, a female:male ratio of 1:1.3; 95% were of northern European (white) origin, 5% originated from the Indian subcontinent, and 8% were the offspring of consanguineous unions. Table 1 summarises the principal findings in this population. Table 2illustrates the distribution of ages at the time of survey. The paucity of patients beyond 40 years of age may reflect underdiagnosis in past years or an increased mortality rate.
Summary of survey results
Distribution of ages at the time of survey
The mean birth weight was 3370 g (range 2050-5400 g). Most pregnancies were uncomplicated, and there was no significant increase in pre- or postmaturity.
AGE AT DIAGNOSIS
The average age at diagnosis was 9 years, lower than that found in our pilot study, where it averaged 15 years8; however, the average age at which parents first noted abnormalities in their children was 3 years (n=77) giving a mean interval of 6 years between first clinical signs and diagnosis.
MAJOR CRITERIA
Visual disorders
Rod-cone dystrophy (atypical retinitis pigmentosa) had been diagnosed by an ophthalmologist in 102 (93%) patients, while the remaining seven patients were all aged less than 8 years. Parents reported that they first noted night blindness in their children at a mean age of 8.5 years (range 1-34 years). The mean age at which patients were “registered” blind was 15.5 years (range 8-43 years). The onset in BBS patients is therefore earlier than in isolated typical retinitis pigmentosa, and progression is rapid, with a mean of seven years from diagnosis to blindness. Other ocular abnormalities noted included astigmatism, strabismus, cataracts, colour blindness, macular oedema and degeneration, and optic atrophy.
Limb defects
Seventy five subjects (69%) were born with at least one accessory digit; in the majority of cases this was fully formed and bony and situated on the lateral border of the hand or foot. Postaxial polydactyly was present in all four limbs in 23 patients (21%). In nine patients (8%) the extra digit was only present on the hands and in 23 (21%) only on the feet. The 34 patients who did not exhibit polydactyly fulfilled other diagnostic criteria; 16 of these had affected sibs with polydactyly.
Short, broad, stubby fingers and toes were reported in 51 patients (46%). In many, the thumb was placed proximally, there was fifth finger clinodactyly, or there was a prominent “sandal” gap between the first and second toes. Nine patients (8%) showed syndactyly; this was usually partial, most commonly involving the second and third toes.
Height and weight
The mean height in affected males was 1.73 m (SD 0.1) (n=30, range 1.57-1.90 m). This differed significantly from the general population mean of 1.76 m (p<0.001). The postpubertal mean height in affected females was 1.62 m (SD 0.13) (n=26, range 1.27-1.91 m); however, this did not differ significantly from the general population mean of 1.63 m (p>0.05).
Overall, 72% of postpubertal subjects were overweight (BMI >25 kg/m2), according to the WHO body mass index classification (BMI=weight ÷ height2). Fifty two percent had a BMI >30 kg/m2 (defined as obese) and 16% >40 kg/m2(defined as morbidly obese). The mean BMI in affected males was 31.5 kg/m2 (SD 5.7) (n=25, median 30.6 kg/m2, range 21.5-46.4 kg/m2), while in affected females it was 36.6 kg/m2 (SD 8.1) (n=23, median 34.8 kg/m2, range 24.2-52.3 kg/m2). We examined 32 of these patients, all of whom had a truncal and rhizomelic distribution of adipose tissue.
Education
Learning difficulties were reported for 68 (62%) BBS patients. Fifty four (50%) attended a special school because of learning difficulties, visual impairment, or both. Before entering a special school, the majority of pupils were placed in mainstream establishments, where only 35 (32%) were receiving any additional classroom help.
Renal tract abnormalities
Only 26 patients (24%) (of whom 11 (42%) were under 16 years at time of diagnosis) were definitely known to have a renal defect, but this may be explained by the fact that only 57 of the patients surveyed (52%) had undergone any radiological investigation of the renal tract (35% had an ultrasound scan, 40% had isotope renography, and 14% an intravenous pyelogram). Of the 57 patients who had been investigated, 26 (46%) were found to have renal abnormalities. Structural abnormalities included renal parenchymal cysts/communicating calyceal cysts (6/57, 10%), calyceal clubbing and blunting (6/57, 10%), fetal lobulation (7/26, 12%), scarring (7/57, 12%), dysplastic kidneys (3/57, 5%), unilateral agenesis (2/57, 4%), renal calculi (1/57, 2%), vesicoureteric reflux with pyelonephritis (5/57, 9%), bladder obstruction (2/57, 4%), hydronephrosis (without bladder involvement) (2/57, 4%), horseshoe kidney (1/57, 2%), and ectopic kidney (1/57, 2%). Six patients (5%) (four of them children, 66%) had chronic renal failure (based on raised plasma urea and creatinine levels) and four (4%) (two of them children) received a kidney transplant. Detrusor/bladder instability, requiring either continuous or intermittent self-catherisation to drain residual bladder urine, was reported in 11 patients (10%).
OTHER FEATURES
Developmental delay
Fifty five patients (50%) were developmentally delayed and late in reaching milestones. More specifically, 46 (42%) showed a delay in walking of up to one year and speech was delayed by up to two years in 47%. Thirty four patients (31%), all males, were slow in passing through puberty.
Neurological and motor defects
Neurological examination of four patients from two families showed moderate hypotonia, with ligamentous laxity, genu valgum, and joint pain. Two further patients had a slowly progressive spastic paraparesis, but polydactyly was present, thereby excluding the diagnosis of Laurence-Moon syndrome.
Forty three patients (40%) showed signs of ataxia and poor coordination. BBS children seem to be clumsy and young adults have very poor balance, particularly on tandem walking and Fogg tests. Dysdiadochokinesia and past-pointing are frequent (79%), both of which appear to be unrelated to reduced visual acuity. There is no evidence of any peripheral sensory disturbance nor loss of joint position sense.
An abnormal gait was reported in 36 subjects (33%), which we found to be broad based in 22 patients examined.
Behaviour
One third of parents/relatives commented on difficult behaviour in young BBS patients, with certain traits continuing into adulthood. These included emotional immaturity, frequent volatile outbursts, and poor reasoning. We have observed inappropriate affect in approximately half the patients and some appear disinhibited. Two patients (2%) in this survey displayed obsessive or obsessive-compulsive behaviour, with frequent hand washing or panic attacks, while five patients (5%) had a history of depression and two (2%) had been diagnosed with schizophrenia. We have noted some BBS children (no figures available) had a tendency to avoid direct gaze and many had difficulty in appreciating abstract thought. All BBS children preferred fixed routines and disliked any deviation from them.
Speech deficit
Speech delay/deficit, requiring speech therapy, was reported in 59 patients (54%). From survey responses and from a specialist speech and language assessment of four BBS children, vocal and speech defects are the main components of this deficit. The voice is high pitched, of breathy quality, and shows poor volume control, while oral and palatal movements appear uncoordinated. Speech is hypernasal and slow, with multiple misarticulations, substitutions (particularly of the first consonant of a word), and omissions of the last syllable of a word. Language and comprehension difficulties are also apparent; these BBS children were unable to repeat sentences accurately and had difficulty interpreting language subtleties correctly.
Hearing
Hearing loss was reported in 23 patients (21%). In 20 cases this was conductive, and associated with chronic otitis media (“glue ear”), but had largely resolved by puberty. However, three patients had unexplained sensorineural hearing loss.
Dental anomalies
Dental problems appear to be common; 29 patients (27%) reported malocclusion, or crowding of the teeth, or had required dental extractions. We observed enamel hypoplasia with yellow discoloration, crowding of teeth, and mild micrognathia. Forty out of 45 patients examined by us (89%) had a high arched palate.
Asthma
We have previously noted childhood onset asthma in a high proportion of BBS patients,8 and in this survey it was reported in 28 patients (25%). Asthma was diagnosed if bronchodilators were used regularly, with benefit. Although the asthma was nearly always of early onset, there was no clear evidence for atopy when patients/respondents were questioned further.
Facial features
Photographs of 70% of the participants in the survey were received. There was considerable, inconsistent, dysmorphism, but a subgroup of patients showed facial similarities comprising deep set eyes, hypertelorism with downward slanting palpebral fissures, a flat nasal bridge with anteverted nares and prominent nasolabial folds, a long philtrum, and a thin upper lip (fig 1). Many BBS patients have a prominent forehead, while adult males show early balding, giving them the appearance of being much older than their chronological age (fig2).

Facial appearance of children with Bardet-Biedl syndrome.

Appearance of adult patients with Bardet-Biedl syndrome (ages from left to right 34, 38, 37, 30, 33, 63, 43 years respectively). Note the frontal balding.
Hypogonadism
All adults examined showed secondary sexual characteristics. In males, hypogonadism was almost universal (being present in 60 of 62 respondents), while maldescended testes were reported by eight patients (13%). The penis was small and often buried in adipose tissue. In females, the mean age of menarche was 13.8 years. Following the onset of menstruation, irregular cycles were reported for the majority of females. Three affected women (2.7%) have given birth to normal, healthy children.
Heart defects
Congenital heart defects were recorded in eight patients (7%). These included, in order of frequency, aortic stenosis (3), patent ductus arteriosus (3), and unspecified cardiomyopathy (2).
Diabetes mellitus
Seven patients (6%) had non-insulin dependent diabetes (NIDDM). Since only a minority of patients surveyed had undergone a fasting glucose measurement or glucose tolerance test, many BBS patients may have undetected NIDDM or impaired glucose tolerance.
Table 3 details other, miscellaneous findings.
Miscellaneous findings (n=109, number affected in brackets)
RELATIVES
Obesity
It has been suggested previously that obesity is an indication of obligate carrier status in BBS.11 12 We found no excess of obesity among carrier parents (fig 3).
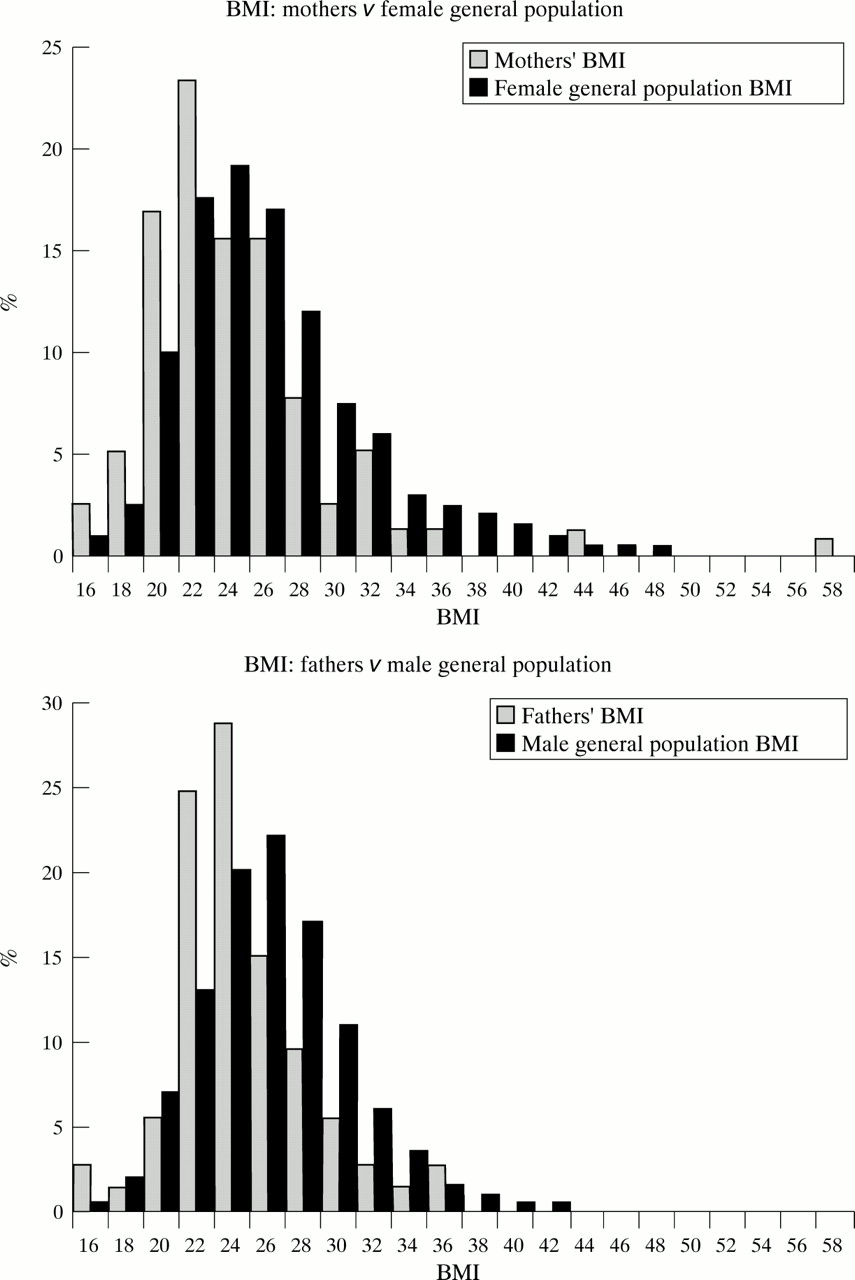
{kind=link}
{kind=link}
{kind=link}
Graph of parents’ BMI values compared with general population.
Renal anomalies
We found that five of 123 sibs (4.1%) of the 109 BBS patients surveyed had one or more congenital renal malformations. Two (1.6%) had unilateral renal agenesis and one also had a complex malformation of the solitary urinary tract. In addition, three (2.4%) showed vesicoureteric reflux. One parent of a BBS patient had unilateral renal agenesis, with a solitary kidney in end stage renal failure, and a second parent (a 37 year old male) had a duplicated renal pelvis and ureter. This man, a second male aged 52 years, and a female aged 40 years, all white parents of affected subjects, had renal cell adenocarcinomas of the same clear cell histopathological type.
Discussion
A Medline search showed that since 1966, 325 articles specifically about the Laurence-Moon and Bardet-Biedl syndromes have been published. Before this, some 400 cases had been published. These syndromes have been reviewed by Green et al,1Schachat and Maumenee,10 and Klein and Ammann.13 Bell14 reviewed a large population based on 273 published pedigrees. We present the largest and most comprehensive survey of living patients with Bardet-Biedl syndrome. A comparison of the cardinal features in each of these reviews is presented in table 4.
Frequency of cardinal signs in the Bardet-Biedl syndrome by study
SEX RATIO
In this study, the male to female ratio was 1.3:1. A similar ratio was found in other studies,13-15 except for that of Greenet al 1 who found a small excess of females. Of the 462 BBS subjects detailed in these studies (table4), 264 were male and 198 female giving a male excess of 1.3. Although all these studies point to an autosomal recessive mode of inheritance there does appear to be an imbalance in the sex ratio which is not entirely attributable to ascertainment bias.
CONSANGUINITY
The consanguinity rate in our study is low (8%) compared with that in other studies,1 13 14 but not surprising given the social structure of the UK population. However, it may also be the result, in part, of a reluctance to admit to, or a lack of awareness of, intrafamily marriage. Bell14 (39%), Klein and Ammann13 (48%), and Green et al 1 (35%) recorded much higher rates. The latter two studies, from Switzerland and Newfoundland, were confined to isolated populations in which consanguinity was more common and the founder effect plays a major role.
DIAGNOSIS
There is clearly a major delay in diagnosing BBS in young patients. This is probably because of both a lack of medical awareness of the condition and the difficulty of making a firm diagnosis owing to the slow emergence of features such as retinitis pigmentosa and renal dysfunction.
EYE DISEASE
The presence of rod-cone dystrophy is the clinical finding most likely to lead to a successful diagnosis. Previous reviews (table 4) all cite a high frequency of retinal dystrophy, thus weighting this sign disproportionately and many cases of BBS remain undiagnosed in its absence, a diagnostic problem which will only be resolved by the development of a biochemical or a direct molecular test.
When compared with hereditary retinitis pigmentosa, the visual impairment in BBS is consistently early in onset; almost all patients (98%) have suffered visual loss before the third decade. Klein and Ammann13 also observed this age relationship; 73% of their population were totally blind by 20 years and 86% by 30 years, while all cases had a visual deficit by this age. We have previously suggested a genetic locus effect on the rate of progression of retinal dystrophy.8
Up to 5 years of age, electroretinograms (ERG) and visually evoked responses (VER) are often normal, but 90% of children will have an attenuated ERG response by 10 years (D Calver, personal communication). Many children develop myopia or strabismus well before retinal changes are apparent, and these should alert the clinician to the possible diagnosis, in conjunction with the other cardinal signs.
LIMB DEFECTS
Polydactyly was observed in 69% of BBS patients and is a useful diagnostic sign, but it may not be present in every case, even within the same family. We observed polydactylous toes three times as often as polydactylous fingers, as did Green et al 1 and Klein and Ammann.13 Riiseet al 16 found polydactyly to be present at birth in 20 of 44 BBS patients (45%). Interestingly, we have observed monozygous male twins with BBS, where one twin was born with postaxial polydactyly of three limbs, while the other had no polydactyly. This suggests that complex influences operate on limb differentiation, despite an identical genotype and a similar intrauterine environment. The presence of brachydactyly is largely a subjective assessment, and its true frequency can only be determined by anthropometric measurements.17
OBESITY
The lower incidence of obesity in our study compared with other reviews (table 4) can probably be explained by the use of different measurement parameters. We used the WHO classification based on body mass index (BMI), where the height is incorporated into the calculation. According to this formula, the BMI of a normal female on the 50th centile for height will be 26 kg/m2 at the 91st centile and 29 kg/m2at the 98th centile. Females above the 91st centile are classed as overweight and above the 98th as obese. Klein and Ammann13 based their obesity estimates on weight alone, and plotted them on age related normal population charts, while classifying any patient over the 50th centile as obese. Greenet al 1 used height adjusted weight charts in a study of 25 BBS subjects, and defined all those above the 90th centile as overweight and all above the 95th as grossly obese.
HEIGHT
Although the mean height of subjects did not differ significantly from the population mean, the range was very wide. We have previously suggested a gene locus effect on height which might account for the variation seen in this cohort.8
EDUCATION
Of those subjects with learning difficulties (62%), half received educational support at a “special school”. The severity of learning disability is usually mild to moderate, with a minority having severe mental retardation.8 Several subjects went on to further education and three completed university degrees. This is similar to findings by Green et al,1 where formal IQ assessments were made, and Riise et al.16 Unusually, several of the respondents reported a talent for mathematics and mental arithmetic while many are reported to have very poor short term memory but excellent long term recall particularly for minutiae.
RENAL AND URINARY TRACT ABNORMALITIES
Renal failure is the major cause of morbidity and early mortality in BBS subjects.18 A wide range of renal abnormalities has been described, with one study2 showing 100% of subjects affected. These were the same patients as described by Greenet al,1 and the high frequency may perhaps represent a sampling bias, since they were ascertained through a nephrology department. Characteristic cystic tubular disease, lower urinary tract malformations, chronic glomerulonephritis, and defects of tubular concentrating ability are among the commonest causes of renal impairment.18 Our study highlights the fact that renal abnormalities are diagnosed in relatively few cases (46%), partly because of the limited investigations performed. In our survey, 18% of subjects investigated had an ultrasound scan (which will not detect the commonest calyceal malformations). We recommend a combination of renal ultrasound scanning and intravenous urography. Despite the presence of underlying renal malformations, only a small number of BBS patients were symptomatic at the time of the survey. It would be of interest to follow a cohort with structural changes present at birth.
In a smaller study (n=41), we found that 33% of subjects had polyuria and polydipsia in the absence of diabetes mellitus.8 This probably represents an underlying nephrogenic diabetes insipidus, which has been well documented.2 This is a useful diagnostic question, particularly when directed at parents. Ten percent of BBS subjects reported problems of incomplete bladder emptying and overflow incontinence, probably owing to bladder instability. This might, however, be neurogenic in origin, secondary to a spinal canal lesion, as these subjects all exhibit gait abnormalities, with varying degrees of lower limb spasticity.19 Further spinal cord imaging studies in this subgroup are warranted. Conversely, bladder anomalies may reflect a more generalised primary defect in the urinary tract.
NEUROLOGICAL ABNORMALITIES
The overlap between Bardet-Biedl syndrome and Laurence-Moon syndrome has been described before.19-22 We believe that the two clinical entities may represent allelic forms of the same condition and that perhaps the older term, Laurence-Moon-Bardet-Biedl syndrome should be reinstated. Moreover, perhaps a more descriptive term such as the polydactyly-obesity-kidney-eye syndrome could be adopted.
Ataxia
Ataxia has been documented rarely in Bardet-Biedl syndrome but is associated with the Laurence-Moon syndrome along with long tract signs.3 23 Many subjects (and their relatives) complain of clumsiness, incoordination, and poor balance and cite the latter as their main mobility problem, far outweighing the inconveniences of blindness. The lower limbs appear to be more severely affected than the upper limbs, which may in part be because of the weight on the lower limbs exaggerating the signs or a structural deformation of the spinocerebellar tracts. Ten patients who have had CT/MRI of the brain failed to show an isolated structural abnormality of the cerebellum which might account for these findings.
BEHAVIOUR
Parents often report behavioural difficulties in childhood with labile emotional outbursts, frustration, and inflexibility, traits which have been documented twice.1 13 Many prefer a fixed routine and tolerate poorly any deviation from it. A few show compulsive/obsessive tendencies. Some children are hyperactive with attention deficit, while others are docile with unwavering attention toward subjects that interest them. Adult patients are often disinhibited and appear unable to recognise social cues.
SPEECH DEFICIT
We have previously reported a high frequency of speech deficit which has been poorly characterised to date.8 The quality of the voice is breathy and often high pitched and nasal. Substitution of the first consonant of a word often leads to incomprehension. The last consonant may also be dropped resulting in incomplete word formation. Language use is occasionally a problem and vocabulary may be limited by learning difficulties. While the pattern of speech, voice, and language disorder is not specific to BBS, it does appear to be consistent and this may be helpful diagnostically, especially in children.
DENTAL ANOMALIES
Dental anomalies in BBS were first documented in 1960.24 More recently they have been described by Kobrinet al,25 Lofterodet al,26 and Borgstromet al.27 The most significant findings were hypodontia, small teeth, enamel hypoplasia, short roots, and a thickened mandibular body. Our finding of high arched palate has not been noted to be a significant feature of BBS before, but is a useful minor sign.
ASTHMA
We have reported asthma previously in 24% of subjects, all of whom were linked to the BBS1 locus (chromosome 11q13).8This is confirmed in the current study (25%) representing a three fold increase on the national prevalence rates (7%), but is unlikely to be the result of a direct association with the α subunit of the high affinity IgE receptor gene mapping nearby, as it is some 6-8 cM away.28 The finding may be because of coinheritance of the two genes, a further asthma locus, or perhaps BBS1 itself plays a role in bronchial hypersensitivity.
FERTILITY
This is more difficult to determine in females than males, and thorough radiological and endocrinological investigations may be indicated. In addition, several genitourinary malformations have been reported.29 30 Males are usually infertile owing to primary gonadal failure; however, we know two males who are fertile and have children. Endocrinological assessments and semen analysis may be helpful.
DIABETES MELLITUS
Non-insulin dependent diabetes mellitus has been described in 45% of patients with BBS,1 but more particularly in Alstrom syndrome (recently mapped to 2p12) in which there is cone-rod dystrophy, obesity, sensorineural deafness, acanthosis nigricans, and diabetes but no polydactyly.31 This should be considered as a differential diagnosis. NIDDM in BBS is a consequence of severe insulin resistance (unpublished data).
MISCELLANEOUS FINDINGS (TABLE 3)
Ten patients, of whom eight were female, had multiple widespread pigmented naevi. Hirschsprung’s aganglionosis has been reported elsewhere.32-34 Several patients had marked joint laxity resulting in multiple dislocations of the shoulder or patella. Two families each had two affected sibs with joint laxity and early onset, lower limb osteoarthritis.
RELATIVES
Obesity
The frequency of obese and severely obese parents did not differ significantly from that of the general population in contrast with Croft et al,12 who found an excess of severely overweight fathers (BMI >31 kg/m2) in their study of 33 patients. They also went on to propose that 2.9% of severely overweight white males might carry a BBS gene.
Renal anomalies
Renal cell carcinoma is an uncommon tumour especially in the age group we have documented (37-52 years). The cumulative risk of developing RCC under 55 years in the general population is 1 in 1041.35 Three cases out of 180 (1 in 60) represents a 17 fold increased risk. Each case was a parent (by definition an obligate carrier of a BBS gene) with the same histopathological picture and one had an accompanying ureteral duplication. Heterozygous carrier effects have been suggested for autosomal recessive conditions such as ataxia telangiectasia where there appears to be a 3-4 fold risk of breast cancer in gene carriers.36 Croft and Swift11have suggested that carriers of a Bardet-Biedl gene have an increased incidence of obesity, hypertension, and renal disease. The high incidence of renal dysgenesis/agenesis is intriguing. The population incidence of unilateral renal agenesis is 0.1%,37 so our finding of 1.6% represents a 16 fold increase; however, the aetiology and incidence of these anomalies are complex and only the emergence of further cases, or identification of gene mutations, will unravel the processes. On the basis of these findings we suggest obligate gene carriers and sibs could be screened for kidney tumours and an ultrasound scan or intravenous urography could be offered to possible carriers to exclude congenital renal tract abnormalities. These renal anomalies and cancer are the subject of further genetic analysis.
NEW DIAGNOSTIC CRITERIA
A new scheme (table 5) for diagnosing BBS is proposed, which should be helpful particularly in children. We are mindful of the difficulty in differentiating between Laurence-Moon syndrome and Bardet-Biedl syndrome using these criteria, but we feel it is important to identify the many overlapping cases highlighted by this study.
Modified diagnostic criteria
CLINICAL SURVEILLANCE
We recommend the scheme in table 6 for initial investigation and follow up of BBS patients. In addition, partners and sibs could be screened for renal malformations and carcinoma.
Initial investigation and follow up recommendations for a person with Bardet-Biedl syndrome
Summary
The results of this comprehensive study show a wide range of previously undescribed features. Not all patients are obese at the time of measurement (72% obese) and polydactyly is present in 69%, so its absence should not exclude diagnosis. In contrast, rod-cone dystrophy is a universal finding among all adults diagnosed with BBS, but symptoms do not begin until about 8 years and signs are often not visible until the early teens. The diagnosis remains difficult in young patients. There are several other ocular associations such as myopia, strabismus, and cataracts which may help to suggest the diagnosis.
Neurodevelopmental delay is common and features such as hypotonia, dyspraxia, poor balance, and ataxic gait are not infrequent. Speech and behaviour is characteristic but more work is required to define their pattern accurately. Learning difficulties, while present in the majority, are in general mild to moderate.
The characteristic renal tract abnormalities in BBS are helpful diagnostically, but are not very frequent.
Dental anomalies, reported in a third of respondents, such as crowding of the teeth, hypodontia, and high arched palate should also be sought.
The presence of a typical facies with “moon face”, frontal premature balding, enophthalmos, and downward slanting palpebral fissures is not necessarily helpful in all cases but when present is characteristic.
Finally, the relatively high incidence of renal developmental anomalies and renal cell carcinoma in relatives of BBS patients may be manifestations of the heterozygous state. Furthermore, these genes may be involved in renal development and disruption of one allele may be enough to increase susceptibility to renal dysgenesis or predispose to malignant transformation or both.
Acknowledgments
We are grateful to Ms Elizabeth Manners for help with the manuscript, Dr Sheena Reilly for advice on speech and language, Dr Lucinda Carr and Ms Alison Wisbeach for developmental assessments, and the LMBBS Society for their help and encouragement. PLB is a Medical Research Council Clinical Training Fellow. ASW is supported by a grant from Action Research (S/P/8178). Finally, we thank the patients and relatives who participated in this study.