Article Text

Abstract
Recently much attention has been given to the detection of submicroscopic chromosome rearrangements in patients with idiopathic mental retardation. We have screened 27 subjects with mental retardation and dysmorphic features for such rearrangements using a genetic marker panel screening. The screening was a pilot project using markers from the subtelomeric regions of all 41 chromosome arms. The markers were informative for monosomy in both parents at 366/902 loci (40.6%, 95% confidence interval 37.0-44.2%) in the 22 families where DNA was available from both parents. In two of the 27 subjects, submicroscopic chromosomal aberrations were detected. The first patient had a 5-6 Mb deletion of chromosome 18q and the second patient had a 4 Mb deletion of chromosome 1p. The identification of two deletions in 27 cases gave an aberration frequency of 7.5% without adjustment for marker informativeness (95% confidence interval 1-24%) and an estimated frequency of 18% if marker informativeness for monosomy was taken into account. This frequency is higher than previous estimates of the number of subtelomeric chromosome abnormalities in children with idiopathic mental retardation (5-10%) although the confidence interval is overlapping. Our study suggests that in spite of the low informativeness of this pilot screening, submicroscopic chromosome aberrations may be a common cause of dysmorphic features and mental retardation.
- idiopathic mental retardation
- submicroscopic chromosome rearrangement
- chromosome telomeres
- 1p monosomy
Statistics from Altmetric.com
Mental retardation (defined as an intelligence quotient less than 70) affects 2-3% of the population and yet aetiological factors are found in less than half of patients who undergo evaluation.1-4 One important cause is chromosome rearrangements that result in segmental aneusomy and alter the dosage of developmental genes.5 Chromosome abnormalities have been documented in up to 40% of patients with severe mental retardation and 10% of people with mild mental retardation.1 6 7 Standard microscopic cytogenetic analysis in children with mental retardation is usually performed by visualisation of G banded chromosomes at an ISCN 400-500 band level. However, in recent years, submicroscopic chromosome deletions and rearrangements have been found in subjects who have had apparently normal karyotypes with this level of G banding.8 In some of these cases, a recognisable pattern of malformations suggested that further investigation of a particular area of the genome with higher resolution techniques would be useful (for example, α thalassaemia-mental retardation syndrome,9 10Wolf-Hirschhorn syndrome,11-13 cri du chat syndrome,14 and Miller-Dieker syndrome15). However, phenotype-genotype correlation for most segmental aneusomy disorders is difficult and screening of the genome is usually necessary.
Screening for submicroscopic chromosome rearrangements in patients with mental retardation but without a recognisable pattern of dysmorphic features has focused on terminal rearrangements. This is because practical tools for whole genome screening are not available and rearrangements that delete or duplicate subtelomeric sequences account for more than 50% of all cytogenetically detectable chromosomal abnormalities.16 The clinical features of patients with terminal segmental aneusomy are heterogeneous and submicroscopic deletions have been described in patients with severe mental retardation and dysmorphic features17-20 and in patients with mild mental retardation and a normal facial appearance.17 21 We have screened 27 families using subtelomeric microsatellite markers22 as a pilot project to test the feasibility of screening for deletions and duplications with PCR based markers. We hypothesised that adding dysmorphic features as an entry criterion in addition to mental retardation would increase the yield of a search for submicroscopic aberrations.
Materials and methods
PATIENT SELECTION
Families were offered participation if at least one child had idiopathic mental retardation and a minimum of three dysmorphic features. Chromosome analysis with G banding at a 400−500 band level was apparently normal in all patients before entry into the study. Ethical approval was granted by the Research Ethics Committee of the Manchester Health Authority and the US National Cancer Institute Institutional Review Board and informed consent was obtained from, or on behalf of, all participants.
CASE REPORTS
Family 5
Intrauterine growth retardation was detected at 38 weeks of gestation. Labour was induced and the baby was born by normal delivery with a weight of 1900 g (<3rd centile), length 51 cm (<3rd centile), and head circumference 31.5 cm (<3rd centile). Dysmorphic features were immediately apparent and the baby had small, almond shaped eyes, upward slanting palpebral fissures, low set, dysplastic ears, and a left cleft lip and palate (fig 1). There were rocker bottom feet with deep creases on the soles and prominent heels. Echocardiography showed a moderate atrial septal defect and tricuspid incompetence which resolved spontaneously within the first year of life and a small patent ductus arteriosus which was ligated at 14 months of age. A renal ultrasound showed dilatation of the kidney collecting systems and a chest radiograph showed 11 pairs of ribs with a bifid right 10th rib. Chromosome analysis of both skin and blood was normal in the neonatal period.

Child from family 5 with 1p deletion in the neonatal period. (Photographs reproduced with permission.)
Her developmental progress has been severely delayed and at 6 years of age she was unable to sit independently and had no meaningful sounds. She has had pronounced feeding difficulties and her growth has been severely retarded with minimal weight gain despite insertion of a gastrostomy tube. At 6 years 4 months, weight was 9.6 kg (50th centile for 1 year), length was 94.5 cm (50th centile for 3 years), and head circumference was 45 cm (50th centile for 11 months). Visual impairment was noted from 3 months and at the age of 4 her vision was assessed as 20/600 with a minor degree of myopia in one eye. Examination of the fundi was normal. Audiological testing has shown a moderate bilateral sensorineural hearing loss. Generalised seizures requiring medication have been present from 2 years of age and a CT scan of her head has shown ventricular enlargement and cerebral atrophy. Other problems have included a mild thoracic scoliosis (20°), anal stenosis, chronic constipation, and intestinal obstruction requiring surgery. At 6 years of age her examination showed plagiocephaly and facial asymmetry, a high forehead, a left esotropia, downward slanting palpebral fissures, a broad nasal bridge, a long philtrum, thin lips, and a long, pointed chin.
Family 14
The pregnancy was normal and the baby was born at term by normal delivery with a birth weight of 3400 g (50th centile). Failure to thrive and dysmorphism were noted from the age of 6 months. At 11 years her level of functioning was equivalent to a 5-6 year old child and she has severe developmental delay. Her height and weight have been below the 3rd centile but her head circumference is on the 50th centile. Dysmorphic features have included a prominent, bulbous nose, flat malar bones, prominent lips, and a large mouth with a small chin. She had hyperextensible fingers with fingertip pads and blunting of the finger tips. In the family, the mother has had normal health but two of her three brothers have had open spina bifida (one brother died at 4 months of age and the other has had surgery) and her only sister had spina bifida occulta. A FISH study to exclude Williams syndrome has been negative.
Family 23
Intrauterine growth retardation was detected on prenatal scanning at 24 weeks of gestation. Labour was induced at 38 weeks and the baby was born by normal delivery with a weight of 1800 g (<3rd centile). The neonatal period was complicated by poor weight gain and marked hypotonia. Her development has been severely delayed. She first walked independently at the age of 5 years and at 16 years had no intelligible words. There have been major behavioural problems with self-injury and aggression towards carers. At 15 years (fig 2), height was 131 cm (50th centile for 9 years) and weight was 38.5 kg (50th centile for 11.8 years). Her head circumference at 16 years was 54.0 cm (25th-50th centile). She had hypertelorism, epicanthic folds, a divergent strabismus on raising her eyes, and bilateral ectropion. There was marked midface hypoplasia. She had a small, asymmetrical nose and a short philtrum with absent nasolabial grooves. Her ears were normally formed and sited and her ear canals were of normal diameter. She had downturned corners of her mouth, a high arched palate, and a receding jaw. There was brachydactyly of the fingers and toes with nail hypoplasia and fifth finger clinodactyly but her thumbs were normally positioned. She had Tanner stage 4 breast development but had not started regular menstruation. Other findings have included a small, haemodynamically insignificant atrial septal defect, severe myopia (−5 dioptres) with bilateral choroidoretinal atrophy, and a 50-60 dB hearing loss in the right ear. She had two convulsions at the age of 7 and was treated with short term anticonvulsant therapy. A CT head scan has shown widespread cortical atrophy with moderate enlargement of both cerebral ventricles and prominence of the cortical sulci.

Child from family 23 with 18q deletion at age 15 years.
MICROSATELLITE MARKERS
The marker names, map position, and heterozygosity scores are shown in table 1.22-29 A modification of the method of Biesecker et al 20 was used for these experiments. DNA was extracted from peripheral blood lymphocytes according to standard techniques. PCR reactions using 10-20 ng of DNA from parent(s) and child were set up in separate wells of a 96 well microtitre plate. The reaction mix contained 1.25 mmol/l dGTP, dATP, and dTTP, 15 μmol/l dCTP, 0.15 mCi α32P-dCTP (3000 Ci/mmol), 0.67 μmol/l forward and reverse primer, 50 mmol/l potassium chloride, 10 mmol/l Tris-hydrochloride (pH 8.3), 1.5 mmol/l magnesium chloride, 0.01% gelatin, 0.45 U Taqpolymerase (Cetus Corporation), and distilled water to a final volume of 15 μl. Plates were denatured at 95°C for three minutes before 30 PCR cycles at 95°C for one minute, 55°C for one minute, and 72°C for one minute with a final extension of 10 minutes at 72°C. A total of 20 μl of formamide containing 20 mmol/l EDTA, 10-20 μg bromophenol blue, and 10-20 μg xylene cyanol was added to the wells and products were separated on 6% polyacrylamide and 8 mol/l urea gels (National Diagnostics), and run at 80 W per gel for 2 hours 20 minutes. Gels were transferred to 3MM Whatman paper and exposed to autoradiography film without drying at −80°C for 12-24 hours.
Microsatellite markers for human chromosome telomeres
FISH STUDIES
In situ hybridisation with telomere specific probes18was performed on cultured lymphocytes or lymphoblastoid cells in children from families 3, 4, 5, 6, 7, 9, 10, 11, 14, 20, 23, and 27 according to a modification of the method of Knightet al.23 A chromosome 1 midisatellite probe (D1Z2, Oncor) assigned to 1p36.330 and a centromere probe for chromosome 1 (D1Z5, Oncor) were used according to the manufacturer’s instructions for family 5.
Results
The 41 markers were fully informative for monosomy at 366/902 loci (40.6%, 95% confidence interval 37.0-44.2%) and fully informative for trisomy at 298/902 loci (33.04%, 95% confidence interval 29.9-36.2%) in the 22 families where DNA was available from both parents (total 41 × 22 × 3 genotypes).31Informativeness for monosomy and trisomy for each chromosome is shown in table 1. In addition, we typed 41 markers from mother and child in five families (total 41 × 5 × 2 genotypes). We detected three instances of non-Mendelian inheritance that were consistent with segmental aneusomy.
In family 23, paternal DNA was not available but the child inherited one allele for marker 18q11 that was different in size from both maternal alleles. Additional markers for the long arm of chromosome 18 were typed to confirm this finding (fig 3). D18S879 (5.0 Mb or 24.08 cM from 18qter29) was deleted in the child but she was heterozygous for marker D18S541 (6.3 Mb or 27.81 cM from 18qter29). Based on marker positions from an integrated map of the genome, we estimated the deletion to be 5.0-6.3 Mb (fig 3). FISH with cosmid probe 2050a6 for the 18q telomere showed no signal for one chromosome 18q homologue in the patient (fig 4).18Chromosome analysis with G banding at a 550 band level showed that the abnormal chromosome 18 had additional G pale material at the telomere that was not appreciated on the original study (fig 5). FISH studies with the remaining 40 telomere specific probes18 and whole chromosome painting probes (Cambio) did not indicate the origin of the additional material (data not shown). The mother had a normal karyotype with G banding and two normal 18q telomere signals with cosmid probe 2050a6 (data not shown).

Polymorphic marker analysis for 18q11tel, D18S879, and D18S541 in family 23. C=child’s lane, M=mother’s lane.

FISH study with cosmid probe 2050a6 for 18q telomere (green signal) and P1 probe 52M11 for 18p telomere (red signal) in child from family 23.

Partial karyotype of child 23 showing derivative chromosome 18.
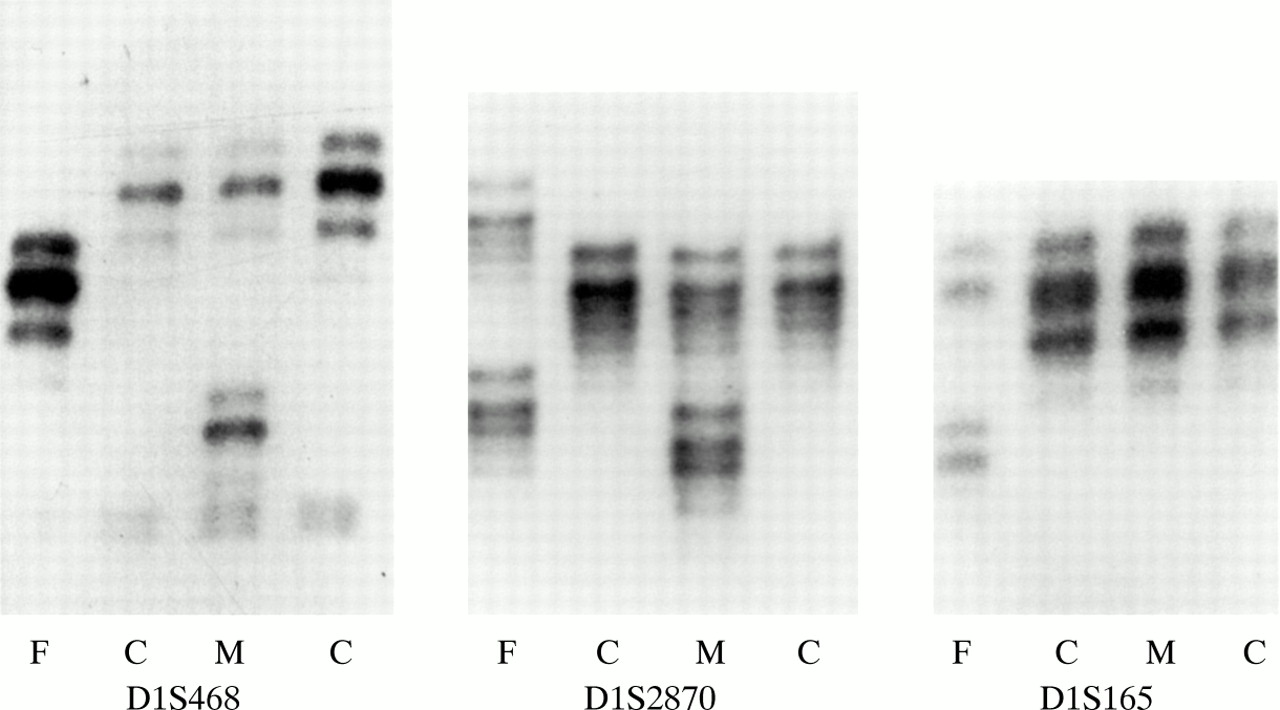
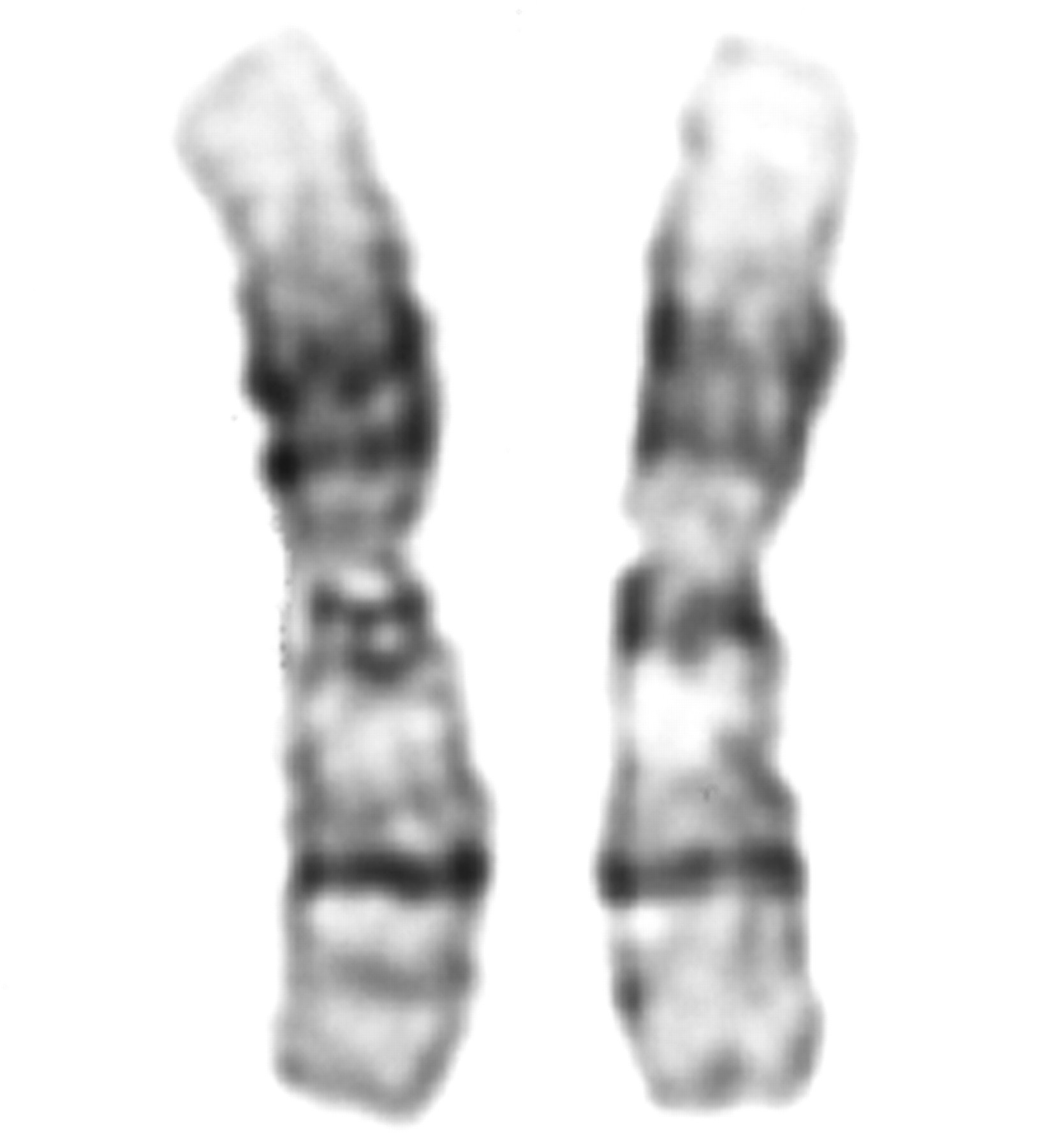
In family 5, the child inherited one maternal allele but no paternal allele with marker D1S468 on chromosome 1p (fig 6). Typing of additional 1p markers refined the breakpoint to a 0.4 Mb region between markers D1S2870 and D1S165 (4.3 Mb and 4.7 Mb from 1p telomere, respectively29). FISH with a midisatellite probe mapped to 1p36.3 (D1Z2, Oncor) confirmed the 1p deletion in the proband (fig 7) and showed normal hybridisation signals on both chromosome 1 homologues in her father (data not shown). The deletion was not visualised with repeat G banded chromosome analysis at 500 band resolution (fig 8).
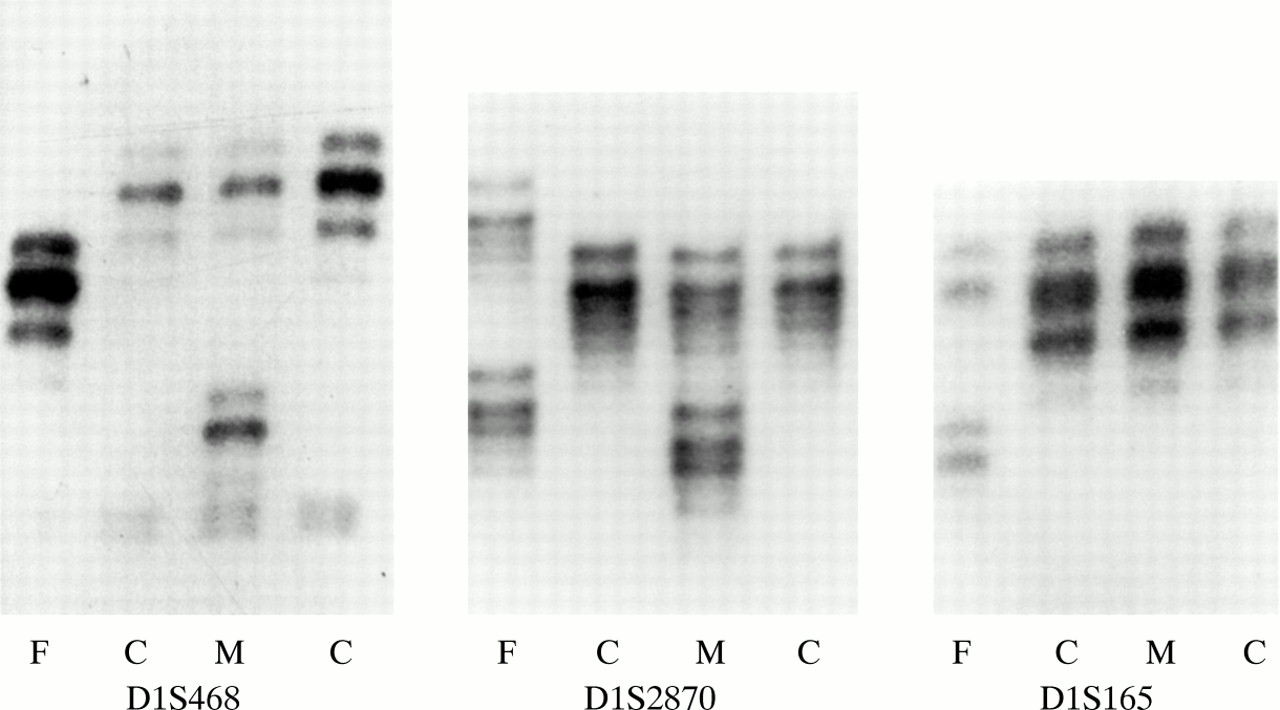
Polymorphic marker analysis for markers D1S468, D1S2870, and D1S165 in family 5. F=father’s lane, C=child’s lane, M=mother’s lane.

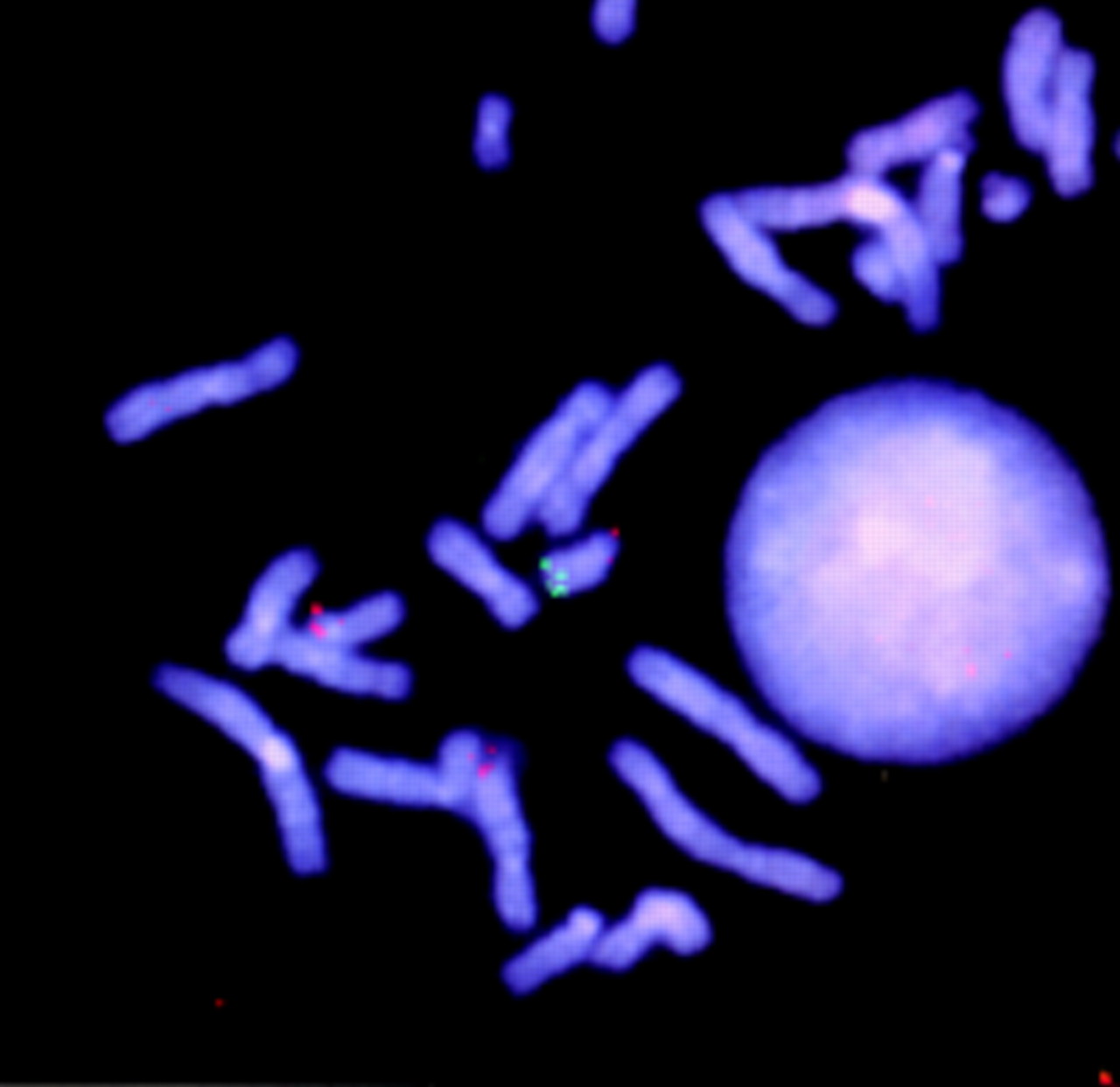
FISH study with probe D1Z2 for 1p36 (green signal) and probe D1Z5 for chromosome 1 centromere (red signal) in child from family 5.

G banded partial karyotype of child from family 5.
In family 14, a maternal allele was absent in the child using marker D7S594 at 7q with different combinations of primers (fig 9). The child was heterozygous for marker D7S2465 (0.61 Mb from the 7q telomere)29 and FISH with cosmid probe 2000a5 for the 7q telomere18 showed two signals (data not shown). The relative positions of the FISH probe and microsatellite marker to 7q telomere are not known and this result could indicate either a small terminal 7q deletion or a polymorphism of the length of the subtelomeric DNA on this chromosome arm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Polymorphic marker analysis for marker D7S594 in family 14. F=father’s lane, C=child’s lane, M=mother’s lane.
Discussion
We report the results of a pilot microsatellite marker screen for duplications and deletions in 27 children with developmental delay and multiple congenital anomalies. Among these 27 patients, two children were diagnosed with newly characterised chromosome aberrations to account for their mental retardation and dysmorphic features. The identification of two deletions in 27 cases gave a detection rate of 7.5% without adjustment for marker informativeness (95% confidence interval 1-24%) and an aberration frequency of 18% if marker informativeness for monosomy was taken into account. The figure of 18% is higher than previous estimates of the frequency of subtelomeric chromosome abnormalities (5-10%) in children with idiopathic mental retardation17 19 although the confidence interval is overlapping.
In spite of the limited informativeness of the marker panel used in this pilot project, microsatellite markers detected two submicroscopic aberrations.17 20 22 The use of genetic marker analysis allows determination of the physical size of deletions and duplications and the parent of origin can be determined. Uniparental disomy can also be detected. However, differences in allele dosage are difficult to detect with markers, limiting the sensitivity of this tool to detect trisomy owing to meiosis I errors or situations where two parents share an allele and the patient is trisomic for another allele and one allele of the same size from each parent. Another limitation of genetic marker screening is that current technical limitations preclude high density screening. We addressed this in the pilot study by limiting our search to the termini of the chromosomes, ignoring the possibility of interstitial aberrations. Future technological advances may allow use of very dense marker panels, which may result in the identification of additional chromosomal aberrations in such populations.
FISH with telomere specific probes has also been used to detect subtelomeric chromosome abnormalities18 19 23 and has the advantage that both balanced and unbalanced rearrangements can be detected. Diagnostic laboratories may also be more familiar with the methods and interpretation of the results. However, further experience is clearly needed before the optimum method(s) for telomere screening can be determined.
The patients in this study were selected on the basis of mental retardation and dysmorphic features, a clinical presentation that is commonly associated with an unbalanced chromosomal abnormality. It is known that patterns of dysmorphic features involving the face, distal limbs, and genitalia can be useful in identifying autosomal chromosomal aberrations.32 The higher yield in this pilot study compared to previous studies of patients with isolated mental retardation suggests that screening for cryptic chromosomal rearrangements may be more useful for patients with mental retardation and dysmorphic features than mental retardation alone.
The range of physical features encountered in patients in this study lends support to the notion that dysmorphic features are important to consider when screening patients for these aberrations. In this study, 12/27 cases had growth retardation (height/weight <3rd centile), six had microcephaly (OFC <3rd centile), and two had relative microcephaly (OFC 2 SD below height/weight). Macrocephaly (OFC >97th centile) was found in three cases. One girl had a moderate sized atrial septal defect and tricuspid incompetence at birth (family 5, see case reports), one child had a small ventricular septal defect, and one child had a small atrial septal defect (family 23, see case reports). Three patients had cerebral atrophy, two others had cerebral atrophy with aplasia or hypoplasia of the corpus callosum, one had an Arnold Chiari malformation, and one child had pachygyria and evidence of abnormal neuronal migration. Three children had orofacial clefting and one had a submucous cleft palate. One child had unilateral renal agenesis (data not shown). Both of the children with newly diagnosed chromosome abnormalities had significant growth retardation and more dysmorphic features than were necessary to qualify for the study. We conclude that future studies should emphasise dysmorphic features when selecting patients to include in panels for chromosomal screening studies.
The child with the de novo 18q deletion had severe mental retardation, growth retardation, hypotonia, seizures, and cerebral atrophy. She had severe myopia with bilateral choroidoretinal atrophy and midface hypoplasia with a narrow, downturned mouth, consistent with 18q deletion syndrome.33-38 Children with “pure” monosomy 1p have a specific syndrome of moderate to severe mental retardation, growth delay, microcephaly, hypotonia, seizures, and visual problems.19 40-48 Common dysmorphic features include a large anterior fontanelle, prominent forehead, deep set eyes, depressed nasal bridge and flat midface, dysplastic or asymmetrical, low set ears, a pointed chin, and fifth finger brachydactyly or clinodactyly, and some patients have cleft lip and palate.41-43 The child from family 5 (see case reports) had similar physical features with severe mental and growth retardation, microcephaly, facial asymmetry, almond shaped palpebral fissures, and poor vision.19 48
In family 14, the absent maternal allele for marker D7S594 in the child (fig 9) with a normal FISH result could indicate either a small deletion of 7q telomere or a telomere polymorphism. The clinical relevance of this finding is unclear. Comparison with previous descriptions of terminal 7q deletions is difficult as reported deletions have been larger in size with more proximal breakpoints or additional cytogenetic imbalance.32 49-52 However, this child does have a bulbous tip to her nose, flat malar bones, and micrognathia as described in other children with terminal 7q deletions.32 49 51 53
These cases highlight important limitations of current clinical genetic practice. Recognition of distinct segmental aneusomy syndromes may be complicated by clinical heterogeneity and some chromosomal aberrations do not apparently cause a uniform phenotype. Because of this difficulty and the limited resolution of G banded cytogenetic techniques, clinicians and their patients may benefit from a tool to perform a broad based screen of the genome for subtle chromosome aberrations. It is highly likely that other “telomere syndromes” will be identified in the near future.
Conclusion
We have screened 27 families with idiopathic mental retardation and dysmorphic features for submicroscopic chromosome rearrangements using microsatellite markers for the 41 chromosome telomeres. The identification of two deletions predicts a yield of at least 18%, which is higher than previously reported detection rates in children with mental retardation without dysmorphic features. We conclude that screening for submicroscopic chromosome anomalies is appropriately directed at children with learning disabilities and dysmorphic features in whom the pattern of physical differences is similar in character and severity to those seen in known segmental aneusomy syndromes.