Article Text

Abstract
Maternal uniparental disomy for chromosome 7 (UPD7) may present with a characteristic phenotype reminiscent of Silver-Russell syndrome (SRS). Previous studies have suggested that approximately 10% of SRS patients have maternal UPD7. We describe a girl with a mos47,XX,+mar/46,XX karyotype associated with the features of SRS. Chromosome painting using a chromosome 7 specific probe pool showed that the small marker was a ring chromosome 7 (r(7)). PCR based microsatellite marker analysis of the patient detected only one maternal allele at each of 16 telomeric loci examined on chromosome 7, but showed both paternal and maternal alleles at four centromeric loci. Considering her mosaic karyotype composed of diploid cells and cells with partial trisomy for 7p13-q11, the allele types obtained at the telomeric loci may reflect the transmission of one maternal allele in duplicate, that is, maternal UPD7 (complete isodisomy or homodisomy 7), whereas those at the centromeric loci were consistent with biparental contribution to the trisomic region. It is most likely that the patient originated in a 46,XX,r(7) zygote, followed by duplication of the maternally derived whole chromosome 7 in an early mitosis, and subsequent loss of the paternally derived ring chromosome 7 in a subset of somatic cells. The cell with 46,XX,r(7) did not survive thereafter because of the monosomy for most of chromosome 7. If the putative SRS gene is imprinted, it can be ruled out from the 7p11-q11 region, because biparental alleles contribute to the region in our patient.
- maternal uniparental disomy
- chromosome 7
- Silver-Russell syndrome
- monosomy duplication
Statistics from Altmetric.com
Uniparental disomy (UPD) is a pair of homologous chromosomes both derived from one parent.1 The contribution of both homologues from a parent to a child is uniparental heterodisomy, while double transmission of one parental homologue to a child is uniparental isodisomy.1 2 UPD is not very rare in man and has been observed for chromosomes 1, 2, 4, 6-11, 13-16, 20-22, and X.2 Maternal or paternal UPD is often associated with a distinct clinical picture. One of the explanations for this parent of origin specific phenotype is genomic imprinting, where only one parental allele is expressed and the other allele is silent (imprinted). Alternatively, it can be explained by homozygosity for a mutated gene. Several possible mechanisms through which UPD arises have been proposed: (1) fusion of a nullisomic gamete with a gamete disomic for the same chromosome (gametic complementation); (2) loss of one member of trisomic chromosomes in a zygote at an early mitotic division (trisomy rescue); or (3) duplication of a monosomic chromosome in a zygote (monosomy duplication or compensation).3 Among them, to our knowledge, only a few cases of monosomy duplication have been reported, occurring in partial trisomies 15 and 21.4-7 There have been 16 cases of maternal UPD for chromosome 7 (UPD7),3 8-15 and at least 12 of them manifested characteristic clinical features reminiscent of Silver-Russell syndrome (SRS).8 11 13-15 Conversely, approximately one-tenth of SRS patients have maternal UPD7.11 Here, we report a SRS patient with mos47,XX,+r(7)/46,XX, where two normal chromosomes 7 show maternal isodisomy.
Patient and methods
PATIENT
The patient, a 20 month old Japanese girl, was born at 34 weeks’ gestation by caesarean section because of premature rupture of the membranes and intrauterine growth retardation (IUGR) with fetal distress. The ages of her mother and father at the time of delivery were both 27 years. There was no family history of any other multiple congenital anomaly/mental retardation syndrome. Apgar scores at one and five minutes were 8 and 9, respectively. The birth weight was 1020 g (−3.0 SD), length 36 cm (−4.4 SD), and occipitofrontal circumference (OFC) 27 cm (−2.0 SD). The patient had poor activity with hypoglycaemia and hyperbilirubinaemia but without respiratory distress. After treatment with glucose and hydrocortisol transfusion and exposure to light, the serum bilirubin level returned to normal within a week but the hypoglycaemia did not improve. Physical examination showed the following abnormalities: triangular face, hypertelorism, micrognathia, clinodactyly of the fifth fingers, trunk and limb asymmetry (fig 1), and patent ductus arteriosus (PDA) that did not resolve with administration of mefenamic acid and indomethacin. The patient was treated with diuretics and water restriction for 46 days. Hoarseness persisted throughout the hospital stay and fibreoptic laryngoscopy examination showed right recurrent nerve paralysis. She was discharged after 105 days with a weight of 2404 g.
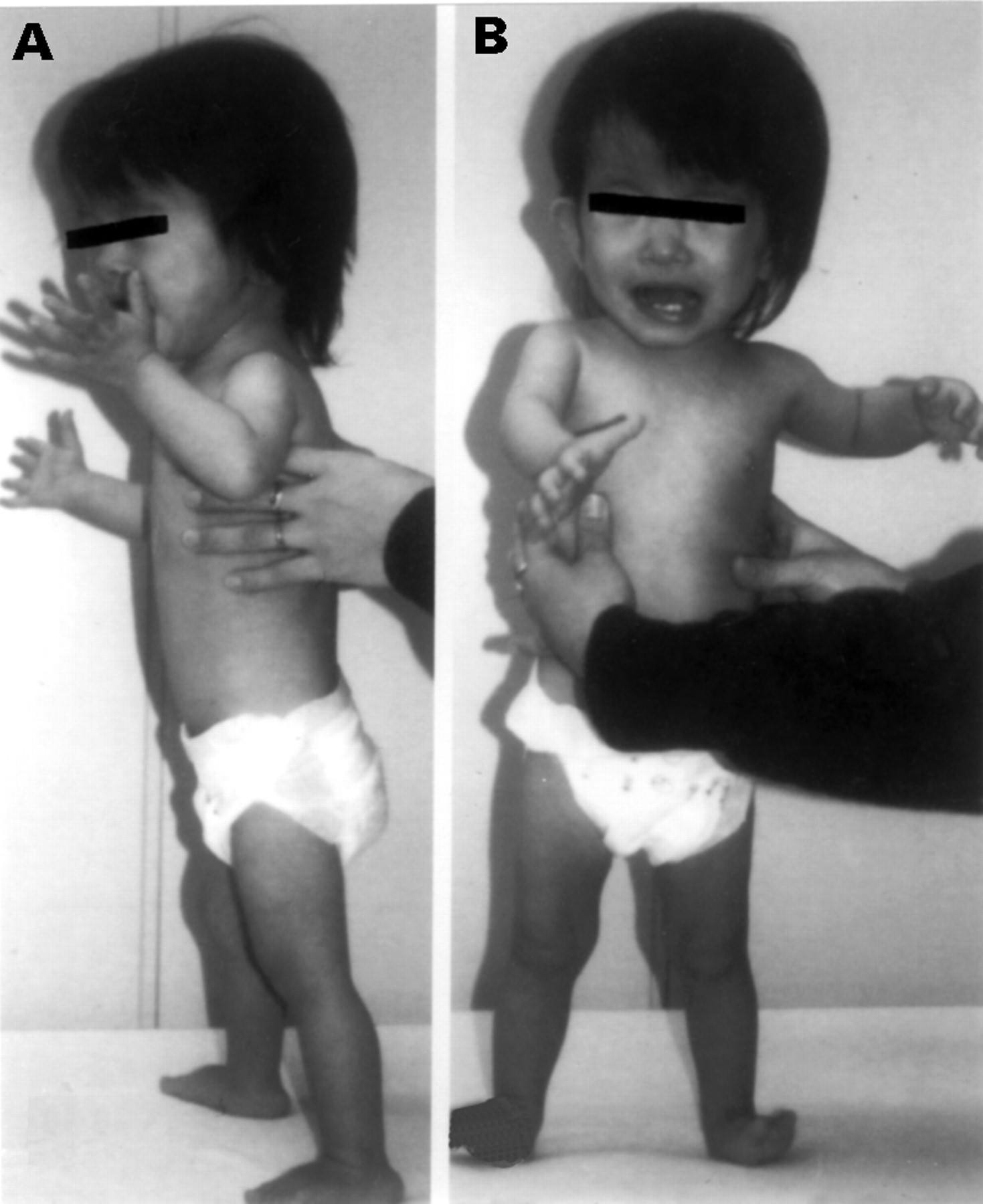
The patient aged 1 year 8 months.
Examination at 18 months of age showed marked growth and developmental retardation with a length of 68.5 cm (−4.7 SD), weight of 5900 g (−3.5 SD), OFC of 44.5 cm (−1.4 SD), and chest circumference of 39.0 cm (−3.7 SD). The OFC was less subnormal than the other growth parameters, indicating top heavy body proportions. The muscles were hypotrophic and bone mineral density showed a low level for her age. These clinical manifestations led to the diagnosis of SRS.
Chromosome analysis on 45 metaphase cells from cultured peripheral blood lymphocytes of the patient showed a 46,XX[30]/47,XX,+r(?)[15] karyotype. Whole chromosome painting using a chromosome 7 specific probe pool (WCP probe, VYSIS, UK) showed that 22 (73%) of 30 cells analysed had two normal chromosomes 7 and eight (27%) had a ring chromosome 7 in addition to the normal chromosome 7 homologues (fig 2). Precise breakpoints on r(7) could not be identified. The karyotypes of the parents were normal.

{kind=link}
{kind=link}
Whole painting FISH on patient’s metaphase chromosomes. Arrows indicate chromosome 7.
ALLELOTYPE ANALYSIS
Parent-child transmission of alleles was studied with CA repeat polymorphic markers located on chromosome 7 (table1).16 17 In addition, markers on chromosomes 2 and 6 were also used to confirm paternity. Genomic DNA was extracted from peripheral blood leucocytes of the patient and her parents. Using Cy-5 labelled primer DNA and the respective unlabelled reverse primers, polymerase chain reaction (PCR) was cycled 30 times under the following conditions: denaturation at 95°C, annealing at 55°C, and extension at 72°C, each for 30 seconds, in a mixture containing 50 mmol/l KCl, 20 mmol/l Tris-HCl (pH 8.5), 1.5 mmol/l MgCl2, 200 mmol/l each of dNTP, and 0.5 U AmpliTaq (Perkin Elmer, USA). Electrophoretic patterns of PCR products were analysed with an automated sequencer (ALFexpressTM, Pharmacia Biotech, Sweden) and software (Fragment ManagerTM, Pharmacia Biotech), and allelotypes of the family members were finally determined, as described elsewhere.18
Allelotypes at CA repeat marker loci in the patient and her parents
Results and discussion
The CA repeat marker analysis indicated that the patient had inherited one maternal allele but lacked any of the paternal alleles at all 16 loci examined from the telomeric regions of chromosome 7 (D7S2563, D7S481, D7S503, D7S526, D7S2497, D7S2422, D7S506, D7S639, D7S657, D7S515, D7S635, D7S530, D7S512, D7S2513, D7S2426, and D7S550) (table 1). However, she had inherited biparental alleles at the four centromeric loci spanning the 7p13-q11 region (D7S2552, D7S499, D7S494, and D7S2503).16 17 Since each of her cell lines, 46,XX and 47,XX,+r(7), contains two normal chromosomes 7, these transmission patterns indicated that she inherited one maternal allele in duplicate at each of the 16 telomeric loci, and one copy of a paternal allele in addition to two copies of a maternal allele at the four centromeric loci. This indicates that the two normal chromosomes 7 were the result of maternal isodisomy and the ring chromosome 7 (r(7)(p13q11)) was of paternal origin. Biparental inheritance of chromosomes 2 and 6 was confirmed at the D2S156 and D6S261 loci.
Most of the clinical features of our patient were consistent with SRS. She was isodisomic for both segments 7pter-p13 and 7q11-qter and trisomic for segment 7p13-q11. To our knowledge, there have been two patients whose trisomic regions overlap that in our patient19 20: a patient with duplications of 7p13-q21 and 5q35-qter had a birth weight of 2900 g, a length of 47 cm, mental retardation, muscular hypotonia, and cat cry,19 while the other patient with duplication of 7p13-p12 had a birth weight of 2895 g, short stature with catch up thereafter (20-50%), failure to thrive, developmental retardation, and clinodactyly.20 Neither patient manifested the SRS phenotype. Thus, it is unlikely that duplication of a 7p13-q21 segment contributes to the cardinal features of SRS, and the same is true for the 7p13-q11 region involved in trisomy in our patient, although the trisomy may have modified her SRS phenotype.
As mentioned above, constitutional UPD may arise through gametic complementation, trisomy rescue, or monosomy duplication. Among them, monosomy duplication may lead to complete isodisomy (homodisomy).1 3 The finding in our patient that there was no evidence of recombination on chromosome 7 in the maternal meiosis (table 1) favours “monosomy duplication” as the mechanism for her UPD7, although the monosomy in the patient is restricted to distal regions (7pter-p13 and q11-qter) of both arms of chromosome 7. The patient may have arisen as a 46,XX,r(7) zygote (partial monosomy for most of chromosome 7), where the normal and the ring chromosomes 7 were of maternal and paternal origin, respectively. Then, duplication of the maternally derived chromosome 7 may have occurred at an early postzygotic cell division, resulting in mosaicism of 47,XX,UPD(7),+r(7)/46,XX,r(7) cell lines. Lethality of the partially monosomic cells and resistance of the UPD cells to cell selection4 5 21 then resulted in a non-mosaic 47,XX,UPD(7),+r(7) karyotype. Finally, the ring chromosome 7 may have been lost in a subset of cells during somatic cell divisions owing to ring chromosome fragility.22 Isodisomy resulting from monosomy duplication has been observed in partial trisomies 15 and 21,4-7 and the UPD7 in our patient provides additional evidence for this phenomenon. Alternatively, a zygote may have had a 47,XX,+r(7) karyotype (partial trisomy 7), where two normal chromosome 7 homologues originated in maternal meiosis II non-disjunction, followed by postzygotic loss of the paternally derived r(7). However, this mechanism is unlikely because we did not observe any maternal heterozygosity at the telomeric region of chromosome 7. Other mechanisms are much less likely, because they need more complex events to explain the chromosome abnormality in our patient.
Maternal UPD7 has been identified in 17 patients including ours.3 8-15 Of these 17, 13 had a common phenotype reminiscent of SRS, such as prenatal and postnatal growth deficiency, characteristic triangular face, short fifth fingers with clinodactyly, limb asymmetry, delayed bone age, and normal mental development.8 11 13-15 It has been suggested that there is at least one maternally imprinted gene on chromosome 7 that conrols intrauterine and postnatal growth.8-15 23 The other four patients had pre/postnatal growth deficiency with or without other features.3 9 10 12 If the putative SRS gene, especially regarding growth deficiency, located on chromosome 7 is maternally imprinted (paternally expressed), it can be ruled out from the centromeric region, 7p13-q11, which corresponds to the remaining extent of the paternally derived ring chromosome in our patient. Similarly, simultaneous occurrence of two isochromosomes for 7p and 7q (maternal UPD(7q) and paternal UPD(7p)) in a previously reported patient24 may also exclude the gene from 7p. The mousePeg1 is a maternally imprinted gene and defects in it result in growth deficiency in the mouse.25Since the human homologue, PEG1, maps to 7q32, a 1 Mb region around the D7S649 locus which is estimated to be located within the deletion in r(7) of our patient (table 1), the deficiency of the gene could contribute to the aetiology of SRS.26 However, a recent report provided evidence against a role of PEG1 in the majority of SRS cases.27 Thus, further efforts to search for the SRS gene are required.
Acknowledgments
We are grateful to Mr Naoki Harada and Ms Hiroko Iwaki for their technical assistance. This work was supported in part by a Grant in Aid for Scientific Research (No 08307019) from the Ministry of Education, Science, Sport and Culture of Japan.