Article Text

Abstract
Relatively few point mutations have been found in the dystrophin gene and of these only two have been associated with mosaicism. A single base insertion has been identified and quantified in a mother of two sons affected with Duchenne muscular dystrophy. It has been determined that she is a somatic mosaic with the mutation present in 25% of lymphocyte DNA. Further tissue lineages have been tested and the time at which the mutation arose was determined to be before the cellular differentiation into the bilaminar disc at approximately eight days after fertilisation. We suggest that the incidence of mosaicism for dystrophin point mutations may be higher than current data suggest.
- Duchenne muscular dystrophy
- point mutation
- mosaic
Statistics from Altmetric.com
Mosaicism may be present in either, or both, the germline and somatic tissues, and in varying proportions depending on the stage of development at which the mutation arose. In the genetic counselling of families with Duchenne muscular dystrophy, the possibility of mosaicism is a major problem, caused by the inability to detect mosaic subjects and uncertainties regarding their frequency.
Duchenne muscular dystrophy (DMD) is a lethal, X linked recessive genetic disease caused by mutations in the dystrophin gene.1 This is a large gene (2.4 Mb, 79 exons) and, possibly because of its size, has a very high mutation rate. Sixty five percent of mutations are large deletions and 5-10% are duplications.2 3 Point mutations are assumed to cause the remaining 25-30% of all DMD cases but relatively few have been found, mainly because of difficulties in detecting these small mutations in such a large gene4 (for details of point mutations see the dystrophin mutation database at http://www.dmd.nl/database.html).
If the dystrophin mutation present in a family has been identified and there is a significant recurrence risk resulting from mosaicism, then it is not essential to be able to quantify the risk accurately, since any offspring or pregnancy can easily be tested for the mutation. However, for families in which the mutation cannot be identified and linkage analysis does not offer a solution, the recurrence risk from mosaicism is often a major factor in the calculation of carrier risks and subsequently influences decisions regarding prenatal testing.
Results of a European collation of data on deletions and duplications suggested that the dystrophin gene exhibits a very high frequency of mosaicism such that an apparent non-carrier mother of a DMD affected male has a risk of approximately 20% of transmitting the mutation to future children if they inherit the high risk haplotype.5There is no information available on the frequency of mosaicism for dystrophin point mutations, and we are aware of only two published cases.6 7 An additional case of mosaicism was presumed to be associated with a dystrophin point mutation because no deletion nor duplication could be detected, but the mutation had not been identified.8 This scarcity of published cases may be a true reflection of a low prevalence of mosaicism for dystrophin point mutations, or may be attributed to the relatively small number of point mutations identified world wide together with difficulties in detecting mosaics. Either way, there is a degree of uncertainty regarding mosaicism when counselling families with a dystrophin point mutation. In this paper we report a third case of mosaicism for a dystrophin point mutation, and we suggest that this phenomenon may not be as rare as it seems.
Methods
The family presented with two boys affected with DMD; their mother has normal serum creatine kinase levels and no previous family history of DMD. The DNA from one of the boys was tested for the presence of a deletion or duplication using fluorescent multiplex PCR assays9 and a slightly larger amplification product was noted for exon 45. Sequencing showed an inserted thymidine in exon 45 (6760^6761insT). Both the mutant and normal exon 45 PCR products were found in lymphocyte DNA from the mother, although the mutant fragment appeared to be at a significantly reduced level as compared to the normal fragment. A specific, quantitative, fluorescent PCR assay was designed to amplify the mutant and normal alleles, allowing complete separation of the 1 bp difference between the normal and mutant PCR products (fig 1). The analysis was carried out on an ABI Prism 310 Genetic Analyzer (Perkin-Elmer, Foster City USA).
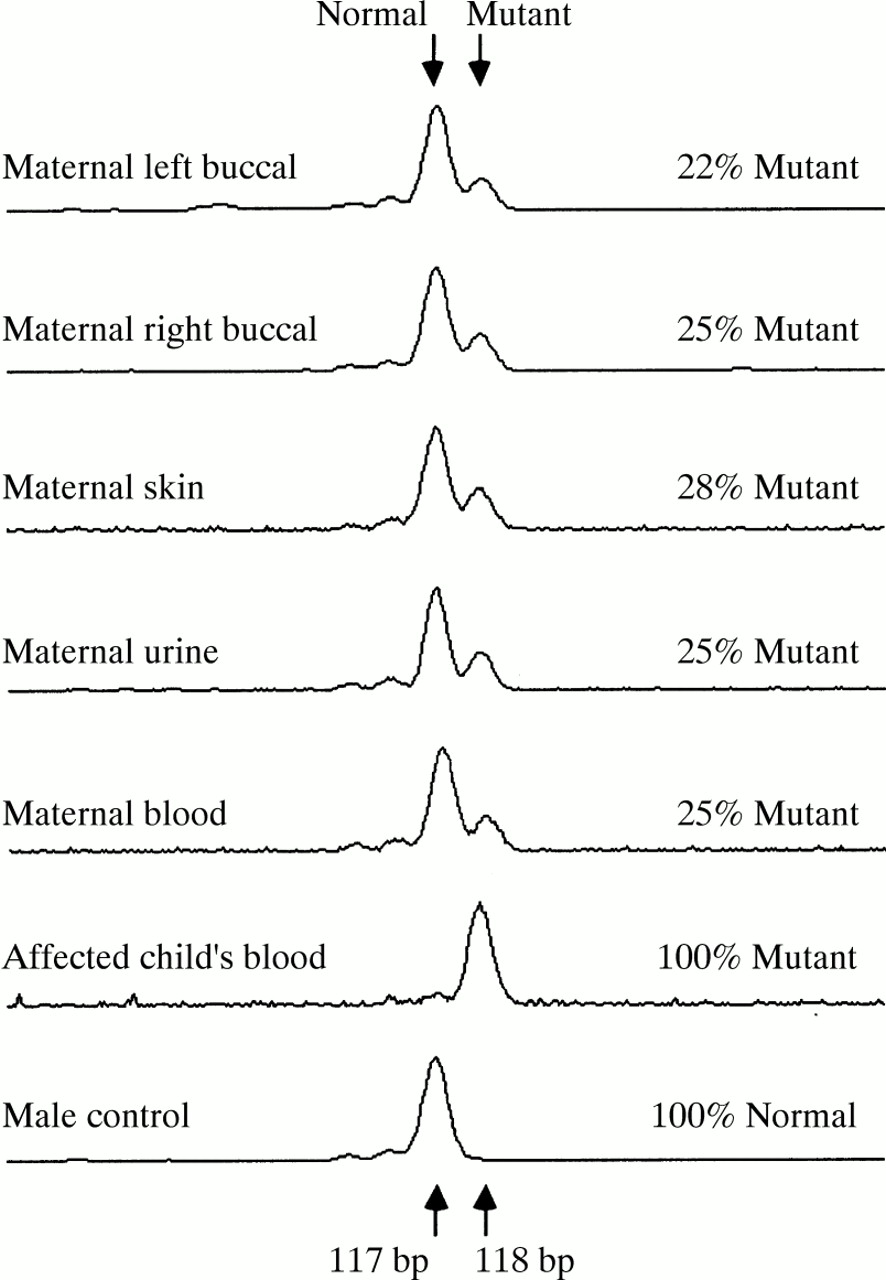
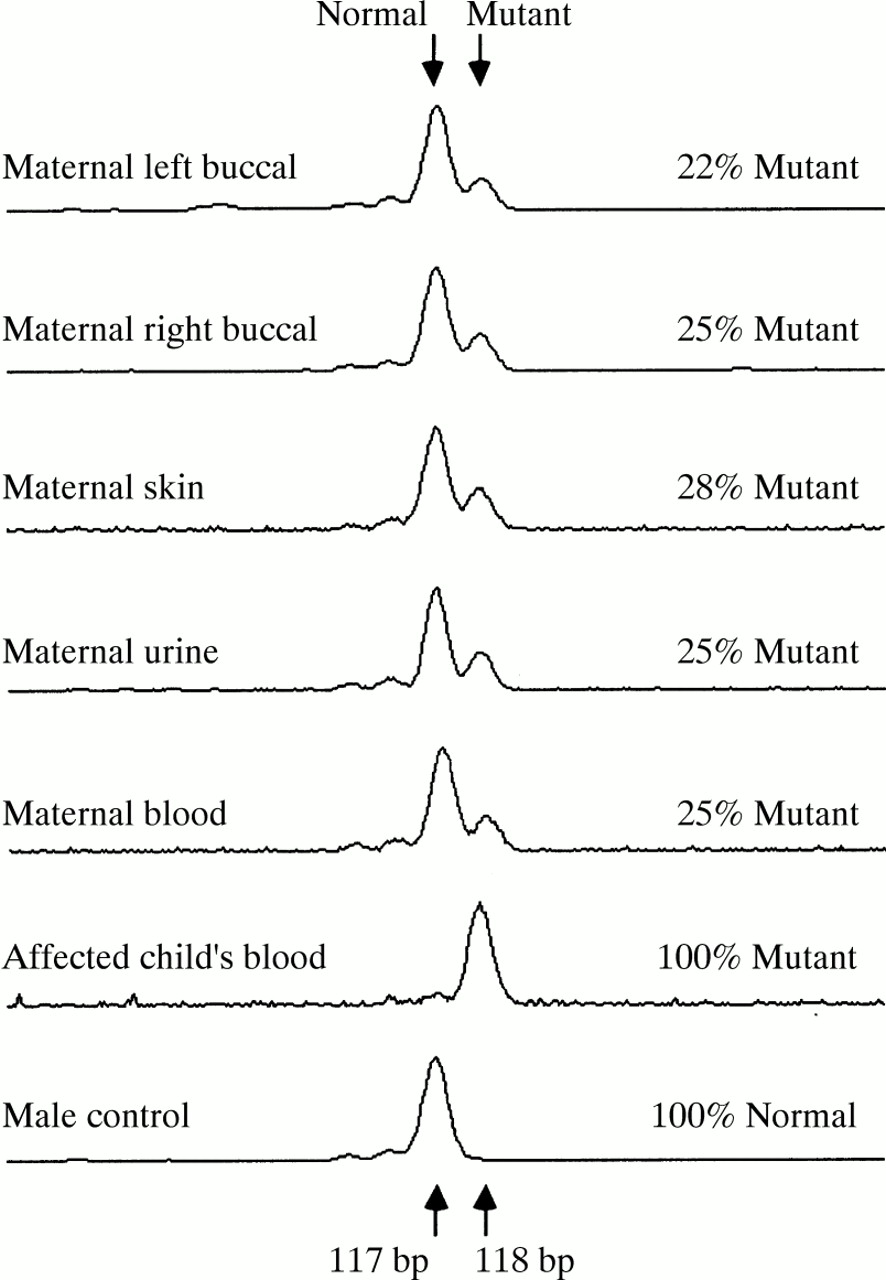
Examples of electrophoretic scans showing peaks produced by Genescan software from the mutant and normal PCR products amplified from various tissues from the mosaic mother, her affected son, and a male control.
A calibration curve was produced by PCR amplification of a series of standards, prepared by diluting total mutant DNA from one of the affected sons to 50%, 40%, 30%, 20%, 10%, and 5%, using DNA from a normal male as the diluent. The relative amounts of mutant and normal amplification products were determined from the areas under their respective peaks on the electrophoretic scans, obtained using GeneScan software (Perkin-Elmer), and plotted against percentage of mutant DNA present in the standards. Linear regression provided a well fitting calibration curve (R2=0.98) and a standard error for predicted values of 2% (fig 2).

{kind=link}
{kind=link}
Calibration curve used to determine the percentage mutant DNA present in different tissues from the mosaic subject.
The mother kindly provided samples from other tissues: a second blood sample, buccal cells (left and right), a full thickness skin biopsy, and urine. The DNA was extracted from the blood using a standard phenol/chloroform method. The buccal and urine samples were centrifuged to pellet the cells (the urine was expected to contain mainly bladder and urethral epithelial cells), digested with proteinase K/sodium dodecyl sulphate (PK/SDS), followed by raised temperature denaturation of the enzyme. These preparations were used in PCR assays without further purification of DNA. The skin biopsy was frozen in liquid nitrogen, pulverised, PK/SDS treated, and then phenol/chloroform extracted. Quantitative PCR amplification and analysis was carried out on all samples using the same techniques as described above. The results were compared against the calibration curve to determine the percentage of mutant DNA present. Each sample was tested 10 times.
Results
The results showed the mutation to be present in 25% (±2%) of peripheral blood DNA, that is, in 50% of cells (table 1). A significantly higher level of mutant DNA was present in the full thickness skin biopsy than in other tissues (p<0.001), and analysis of variance of the other sources showed some evidence of variation (p<0.025). The overall variation is possibly a result of the different tissues originating from separate germ layers. Because the mutation is present in all tissue lineages tested, and in the germline, it can be deduced that the mutation arose before the differentiation of the bilaminar embryonic disc, which occurs at approximately eight days after fertilisation.10
Results of testing for the mutation in different tissue samples from the mosaic subject, showing the percentage mutant DNA detected in 10 repeat tests on each sample, with their mean and standard deviation
Discussion
Since it is not possible to test maternal gonadal tissue, we do not know whether the mother has germline mosaicism or whether all her ova carry the mutation. The mutation is certainly present in a significant frequency as it has been transmitted to both children, and if germline tissues carry the mutation to the same extent as other tissues, only a 25% recurrence risk would be expected.
In the other two published reports of mosaics with dystrophin point mutations, the mutation was present in somatic DNA of one,7 but not the other.6 A recent review of 38 published cases of germline mosaicism in a variety of inherited disorders showed that somatic mosaicism could be detected in approximately half of all cases.11 Somewhat surprisingly, this observation was irrespective of whether the mutation had been detected by Southern blot hybridisation or by the more sensitive method of PCR. However, if the data are divided according to the type of mutation, somatic mosaicism was detected in 12 of 16 cases (75%) that involved a point mutation, in four of seven cases (57%) with a rearrangement, and in only three of 12 cases (25%) where the mutation was a gross deletion. These data could offer an alternative explanation for the small number of mosaics that have been identified with dystrophin point mutations. The majority of the point mutation cases summarised in the report of Zlotogora11 were from autosomal dominant disorders, with the mosaicism being identified because the mutation was present but the expected clinical phenotype was absent. For an X linked disorder such as DMD, if the mother of an affected son is shown to have the mutation somatically, she will be diagnosed as a carrier, rather than a mosaic, unless the ratio between the normal and mutant alleles is significantly different and detectable (as in our case). If a large proportion of mosaics with point mutations are somatic mosaics (75% in the data of Zlotogora11), it is possible that a number of women who have been classed as carriers are in fact undetected mosaics with a high level of mutation present somatically.
If mosaic subjects can be identified somatically in 50% of cases, this implies that if a mutation is not detected at the somatic level, a subject’s risk of germline mosaicism could be reduced by a half. However, this would still leave a significant recurrence risk in the case of DMD, and a means of identifying all mosaics is desperately needed. With the introduction of more sensitive and quantitative methods of mutation detection, a higher proportion of mosaics may be detected in the future, furthering our knowledge of mutational events. On a more practical level it would enable counsellors to give more accurate risks to those families in which mosaicism is currently a problem.
Acknowledgments
We thank the mother of the patient for her cooperation and generous donation of samples, Dr Christine Garrett for arranging and collecting the samples, Dr Cathryn Lewis for help with statistics, Dr Mary Seller for helpful discussions, and Action Research for funding this work.