Article Text

Abstract
Tuberous sclerosis complex is an inherited tumour suppressor syndrome, caused by a mutation in either the TSC1 or TSC2 gene. The disease is characterised by a broad phenotypic spectrum that can include seizures, mental retardation, renal dysfunction, and dermatological abnormalities. The TSC1 gene was recently identified and has 23 exons, spanning 45 kb of genomic DNA, and encoding an 8.6 kb mRNA. After screening all 21 coding exons in our collection of 225 unrelated patients, only 29 small mutations were detected, suggesting that TSC1 mutations are under-represented among TSC patients. Almost all TSC1 mutations were small changes leading to a truncated protein, except for a splice site mutation and two in frame deletions in exon 7 and exon 15. No clear difference was observed in the clinical phenotype of patients with an in frame deletion or a frameshift or nonsense mutation. We found the disease causing mutation in 13% of our unrelated set of TSC patients, with more than half of the mutations clustered in exons 15 and 17, and no obvious under-representation of mutations among sporadic cases. In conclusion, we find no support for a genotype-phenotype correlation for the group of TSC1 patients compared to the overall population of TSC patients.
- tuberous sclerosis complex
- mutations
- genotype-phenotype correlation
Statistics from Altmetric.com
Tuberous sclerosis complex (TSC) is an autosomal dominant neurocutaneous disorder characterised by the growth of hamartomas in many tissues and organs, including brain, skin, heart, and kidney.1 Common neurological manifestations including seizures and mental retardation have their onset during early childhood, while cysts and angiomyolipomas in the kidney mostly become apparent during adult life. Considerable clinical variation is observed between as well as within families.2 At least 60% of TSC patients represent sporadic cases, as they have unaffected parents.3
Linkage analysis has shown locus heterogeneity for TSC, with one locus on chromosome 94 and a second locus on chromosome 16.5 About half of the large families can be linked to the TSC1 locus on chromosome 9q34 and the other half to the TSC2 locus on chromosome 16p13.6 7 The TSC1 and TSC2 genes were identified by positional cloning8 9 and there is abundant evidence that both genes act as tumour suppressor genes.10-13
The TSC2 gene consists of 41 exons, spanning 43 kb of genomic DNA.14 It encodes a 200 kDa protein, tuberin, which has a putative GAP activity for rab515 and rap1,16two members of the ras superfamily of small GTPases. The mutational spectrum of TSC2 includes a number of large deletions often disrupting the PKD1 gene as well,17 18 but also point mutations19-27 and a number of missense changes.28
The TSC1 gene contains 23 exons and encodes an 8.6 kb mRNA. It spans 45 kb of genomic DNA and codes for hamartin, a 1164 amino acid protein of 130 kDa. Analysis of the amino acid sequence showed a potential coiled coil domain at the C-terminus but no homology to tuberin or any other known vertebrate protein was detected.8
The first report describing the molecular genetic and phenotypic analysis of the TSC1 gene suggested that all mutations are small changes, that TSC1 mutations are less common in sporadically affected subjects, and that there is a reduced risk of mental retardation in TSC1 related disease.29 The goal of this study was to construct the mutational spectrum of the TSC1 gene in our collection of TSC patient samples by Southern blot and SSCP analysis. This would enable us to determine whether there is a significant difference in the detection rate between familial and sporadic cases and whether there is a genotype-phenotype correlation for the TSC1 group compared to the overall population of TSC patients.
Patients and methods
PATIENT SELECTION AND DNA ISOLATION
In this study, 225 unrelated patients with tuberous sclerosis complex, diagnosed according to the criteria of Gomez,1were included. Eighty-two patients represented familial cases (36%) with at least one first degree relative. Three large families showed linkage to 9q34.7 One hundred and forty-three patients were designated sporadic cases (64%), in the absence of an apparent family history of TSC. Patients with a TSC2 mutation were excluded from the 225 cases. DNA was isolated from peripheral blood cells according to standard procedures.30 Paternity tests were performed using the Profile kit from Perkin Elmer.
SOUTHERN BLOTTING ANALYSIS
Genomic DNA (6 μg) of 200 unrelated patients was digested with four different restriction enzymes (EcoRI,HindIII, PstI, and TaqI) and run on a 0.7% agarose gel. Southern blotting and hybridisation were performed using standard methods.31 Three cDNA clones were tested on the blots: a 5′RACE clone which had been amplified from a fetal brain cDNA pool (bp 24-1696) (Clontech), an RT-PCR product (bp 1616-3684) generated from fibroblast RNA, and a fetal brain cDNA clone (bp 4100-8600). The three probes cover the coding sequence as well as the 5′ and 3′ untranslated region of the TSC1 gene.
SSCP ANALYSIS AND DNA SEQUENCING
Sequences of primers used for amplification of the 21 coding exons of the TSC1 gene are provided athttp://expmed.bwh.harvard.edu/projects/tsc/. For exon 22, a new intronic forward primer was designed (5′-atactaccagcttactttccata-3′). SSCP analysis was performed according to Orita et al 32 and 2 μl of the PCR product were applied to the Pharmacia GenePhor Electrophoresis system. Gels were run for 2.5 hours at 5°C and 18°C. Running conditions for two gels were 600 V, 50 mA, and 10 W. Subsequently, bands were visualised using a DNA silver staining kit (Pharmacia) in a Hoefer automated gel stainer. Variant patterns were further characterised by direct sequence analysis of the PCR products on an automated DNA sequencer (ABI 377) using the cycle sequencing dye primer kit (Perkin Elmer).
ASO HYBRIDISATION
Oligonucleotides for ASO hybridisation were designed for the mutated and normal sequence. ASO hybridisations were performed at 37°C for 30 minutes. Filters were washed to 0.3 × SSC for 10 minutes at 37°C.
Results
SCREENING FOR LARGE ABNORMALITIES
No large insertions or deletions were identified in the TSC1 gene by Southern analysis in 200 patients. Only in one case (T2965) was an aberrant restriction pattern in a TaqI digest detected (fig 1A), but no consistent change was seen with other enzymes. Comparing the genomic sequence of the TSC1 gene with the size of the extra fragment, we were able to locate the lostTaqI site in exon 15. Sequencing exon 15 of the TSC1 gene of this patient showed a C to T substitution at bp position 1719 (fig 1B), resulting in the nonsense mutation R500X. The presence of this mutation was confirmed by allele specific oligonucleotide (ASO) hybridisation (fig 1C).

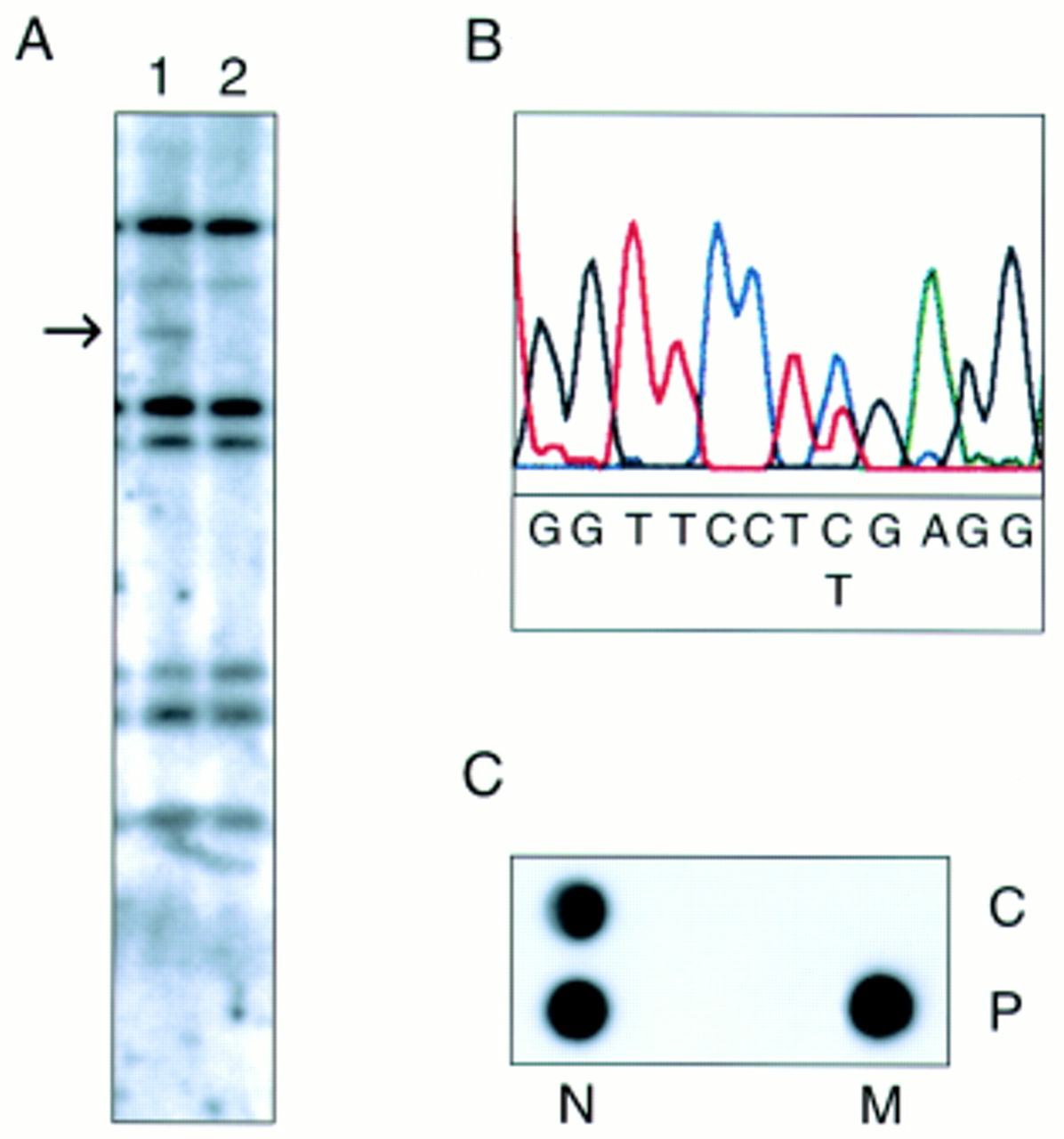{kind=link}
(A) Southern blot of TaqI digested DNA from two unrelated TSC patients, hybridised with a 5′ TSC1 cDNA probe (nt 24-1696). A novel 3 kb fragment is detected in lane 2, indicated by the arrow. (B) Direct sequence analysis shows the de novo nonsense mutation C→T (R500X) in a TaqI site in patient T2965. (C) ASO hybridisation of the R500X mutation in patient T2965 (P) and a control (C). N=normal allele, M=mutant allele.
SCREENING FOR SMALLER MUTATIONS
Systematic SSCP analysis was undertaken to screen the 21 coding exons in the TSC1 gene for small mutations. In total, 25 different mutations were found in 29 subjects out of the set of 225 unrelated patients (table 1). All types of mutations resulting in a truncated protein have been observed: small deletions/insertions, nonsense mutations, and splice site mutations. In addition, eight different missense changes (table 2) were seen in nine unrelated patients.
Mutations identified in the TSC1 gene
Missense and silent changes in the TSC1 gene
Fourteen of the 29 mutations were small deletions, ranging from 1 to 23 bp. Three of these mutations have been reported previously.8 In two patients, we detected in frame deletions of 3 and 9 bp respectively. In family T1298, a 3 bp deletion in exon 7 segregated with the disease phenotype and resulted in a small amino acid change (Asp-Phe to Ile at position 198) in the protein. The grandparents, who had no signs of TSC, tested negative for the mutation. In a sporadic patient (T5913), 9 bp were deleted in exon 15, also leading to a different protein product (Cys-Lys-Ile-Pro to Ser at aa position 586). Both parents tested negative for the mutation. Biological parenthood was confirmed for both in frame deletions. All the other deletions led to a premature stop codon. Nonsense mutations were detected in 11 cases; R692X was present in four sporadic cases and in one family. Three insertions were identified: a single base pair substitution in exon 7 and exon 10 in familial cases, and a duplication of 28 bp in exon 17 in a sporadic patient. A substitution at a splice site (bp postition 432-1) was detected in a sporadic patient, of whom the parents tested negative for the change. The most downstream mutation detected is a 1 bp deletion in exon 20 in the middle of the coiled coil domain of hamartin. No mutations were found 3′ of the coiled coil domain (aa 719-998).
MISSENSE AND SILENT CHANGES
Nine abnormal SSCP patterns were observed representing missense or silent changes (table 2). In four cases the change was also found in the normal population. These polymorphisms were R190S (present in the unaffected parent and absent in the affected parent), E445 (allele frequency of 16% in the normal population), H732Y (allele frequency of 0.5%), and A943. E445, H732Y, and A943 have been reported before.29 None of the additional missense abnormalities have been confirmed to represent the disease causing mutation yet. E51D probably represents a polymorphism, since the amino acid change is in a conserved group. For L191H and M224R, neither parent was available for testing.
TSC1 MUTATIONS IN FAMILIAL AND SPORADIC CASES
Eighty-two of our 225 unrelated patients (36%) had other affected family members. Of the 29 mutations, 13 were identified in the 82 familial TSC patients (16%) and 16 in the 143 sporadic cases (11%). Hence, we found no significant difference in detection rate between familial and sporadic cases. In half of the sporadic cases, both parents were available for analysis and tested negative for the mutation. In the other sporadic patients, DNA of both parents was not available, but there was no clinical indication of TSC disease in the family.
CLINICAL SYMPTOMS VERSUS TYPE OF MUTATION
Patients with a mutation in the TSC1 gene were scored for the most frequent skin, brain, kidney, and heart lesions detected in TSC. A distinction was made between truncating mutations detected in TSC1 (deletion, insertion, splice site, and nonsense mutations) and the in frame deletions in exons 7 and 15 (table 3). Comparing both types of mutations, no obvious correlation could be detected between the genotype and phenotype in the TSC patients. The missense changes were left out of the analysis, because so far none of them has been confirmed to represent a disease causing mutation.
Overall summary of clinical features of all patients with mutations in the TSC1 gene. A distinction has been made between patients with a stop mutation and an in frame deletion
CLINICAL MANIFESTATIONS IN PATIENTS WITH THE RECURRENT MUTATION R692X
The mutation R692X was present in four sporadic TSC patients and in two patients from the same family. The clinical data of these six patients are summarised in table 4. Almost all patients have a history of epilepsy. No renal lesions were detected, but only two patients are older than 15, so these results could be biased by the later onset of cysts and angiomyolipomas. All other symptoms were scored at least once. The patients with the R692X mutation do not share an obviously similar phenotype.
Clinical features of patients with mutation R692X
Discussion
After screening the 21 coding exons of the TSC1 gene, 29 mutations were detected, all of them small changes. The only mutation detected by Southern blot analysis was the substitution of a C for T in aTaqI (TCGA) restriction site, resulting in a stop codon. Since four different restriction enzymes have been used to test a selection of our TSC patients on Southern blots, it is unlikely that large abnormalities disrupting the TSC1 gene have remained undetected. Previous mutation studies in the TSC2 gene have shown a diverse mutational spectrum including large rearrangements, deletions, insertions, and nonsense and missense mutations. In the TSC2 gene, approximately 10% of the mutations detected so far were large deletions, often resulting in disruption of the neighbouring PKD1 gene as well. A possible explanation for the lack of large mutations in TSC1 may be the presence of unknown neighbouring or intragenic genes that are essential for embryonic development and survival. Although all the mutations detected in the TSC1 gene were small, we could not confirm any missense change as the disease causing mutation. Only one missense mutation in TSC1 has been reported before by Joneset al (personal communication), but this de novo change in the gene appeared not to be the disease causing mutation. Conversely, a number of missense mutations have been reported in the TSC2 gene.28 It remains to be explained why the mutational spectrum of the TSC1 and TSC2 genes is different. Despite the differences, most of the mutations in either TSC1 or TSC2 lead to a truncated protein, which is in concordance with a loss of function mechanism. Results obtained by interaction studies indicate that hamartin and tuberin function as a complex,33 which supports the phenotypic overlap observed between TSC patients with either a TSC1 or TSC2 mutation.
So far we have detected a mutation in the TSC1 gene in 13% of our unrelated TSC collection, screening all of the coding region of the gene by SSCP analysis. It is likely that this technique fails to detect all of the mutations and the promoter region has not been tested yet. We only detected the disease causing mutation in two out of three of our families which were clearly linked to chromosome 9. This number is too small to give an indication of the ratio of undetected mutations in the TSC1 gene, but we expect that for the whole group of TSC patients, the majority of the mutations will be found in the TSC2 gene. We did not observe a significantly larger number of TSC1 mutations in our familial cases than in the sporadic population, as was found in a recent study,29 although it is possible that some of our patients were misclassified as sporadic, because in only half of these cases was material from both parents available.
We found a clustering of mutations in exons 15 and 17, in which 14 out of 29 identified mutations were present. The high mutation rate in exon 15 had already been observed when the TSC1 gene was identified8 and can be partially explained by the size of the exon (17% of the coding region). Furthermore, the high proportion of recurrent mutations detected in exons 15 and 17 suggests that part of these exons are particularly prone to nucleotide changes.
The truncating mutations as compared to the in frame deletions in the TSC1 gene did not show an obvious difference in clinical phenotype in the patients, suggesting that there is no clear correlation between the nature of a TSC1 mutation and the clinical phenotype. The six patients with the recurrent mutation R692X also displayed a wide range of clinical symptoms. This is comparable to the clinical differences detected within families with TSC. The phenotypic differences in TSC patients are more likely to be caused by mechanisms such as a second hit,10-12 somatic mosaicism,18 and modifying genes. The latter has also been proposed to contribute to the complex phenotype in the comparable “monogenic” disease neurofibromatosis 1 (NF1).34 In conclusion, we found no support for a different phenotypic spectrum in patients with a mutation in the TSC1 gene compared to the overall population of TSC patients.
Acknowledgments
The first two authors contributed equally to this work. We are grateful to the patients and their families for their participation, to the clinicians and the TSC Working Group Rotterdam for referral of the TSC patients, and to Professor H Galjaard for continuous support. This work was funded by the Dutch Organization for Scientific Research (NWO), the Dutch Praevention Fund (grant number 28-1723), and the Dutch Kidney Foundation (grant number C93.1313).