Article Text

Abstract
A 3 year old boy with a de novo deletion (14)(q11.2q13) of paternal origin encompassing the region from D14S264 to D14S70 is described. The patient presented with severe psychomotor retardation, bilateral cleft lip/palate, bilateral colobomas of the optic nerves and retinas, agenesis of the corpus callosum, pes calcaneovarus, reduced oesophageal peristalsis, and swallowing difficulties. This is the first reported case of PAX9 hemizygosity in humans. Haploinsufficiency of the PAX9 gene might be expected to cause some of the developmental defects and the dysphagia. Another haploinsufficiency candidate gene, the bZIP transcription factor gene NRL, which is specifically expressed in neuronal cells and the eye during embryogenesis, was excluded from the deletion interval.
- chromosome 14
- deletion
- q arm
- PAX9
Statistics from Altmetric.com
Deletions of the proximal long arm of chromosome 14 are very rare. To our knowledge only nine patients, including two sibs and two prenatally diagnosed cases, have been reported to date.1-7 Here we describe clinical and molecular data of a 3 year old boy with an interstitial deletion (14)(q11.2q13).
Case report
This male patient was the second child born to 33 year old parents. There was no family history of recurrent abortions, consanguinity, or mental retardation.
He was born after an uncomplicated pregnancy with a weight of 2500 g, a length of 47 cm, a head circumference of 32 cm, and Apgar scores of 8/9/10.
He was noted to have the following features at birth: bilateral cleft lip/palate, hypertelorism, bilateral colobomas of the optic nerves and retinas, marked micrognathia, apparently low set, large, dysplastic ears, a short neck, and pes calcanovarus. Cranial ultrasound showed agenesis of the corpus callosum and an asymmetrical ventricular system. Laboratory investigations were remarkable only for high TSH levels and hypothyroidism. He was intubated and ventilated for seven days. During the first year, severe psychomotor retardation, microcephaly, marked muscular hypotonia, and cortical blindness were noticed. MRI at 12 months showed, in addition to agenesis of the corpus callosum, a reduced brain volume with normal cortical thickness and normal cortical lamination, suggestive of a decreased number of neurones (lissencephaly type IV, radial microbrain). He had severe fevers, probably because of abnormal central thermoregulation. Cardiac ultrasound showed a persistent foramen ovale (5 mm). EEG and abdominal ultrasound were normal.
Despite surgical correction of the clefts and medical therapy, the patient continued to have eating difficulties with recurrent emesis and swallowing problems resulting in poor weight gain. Sonographic investigations showed reduced oesophageal peristalsis after reflux. At 2 years, a gastrostomy tube was placed and his weight slowly increased. At last examination (3 years), he had a height of 93 cm (3rd centile), a weight of 8.5 kg (<<3rd centile), and a head circumference of 42 cm (<<3rd centile) (fig 1). He had severe hypotonia and was unable to grasp or sit.
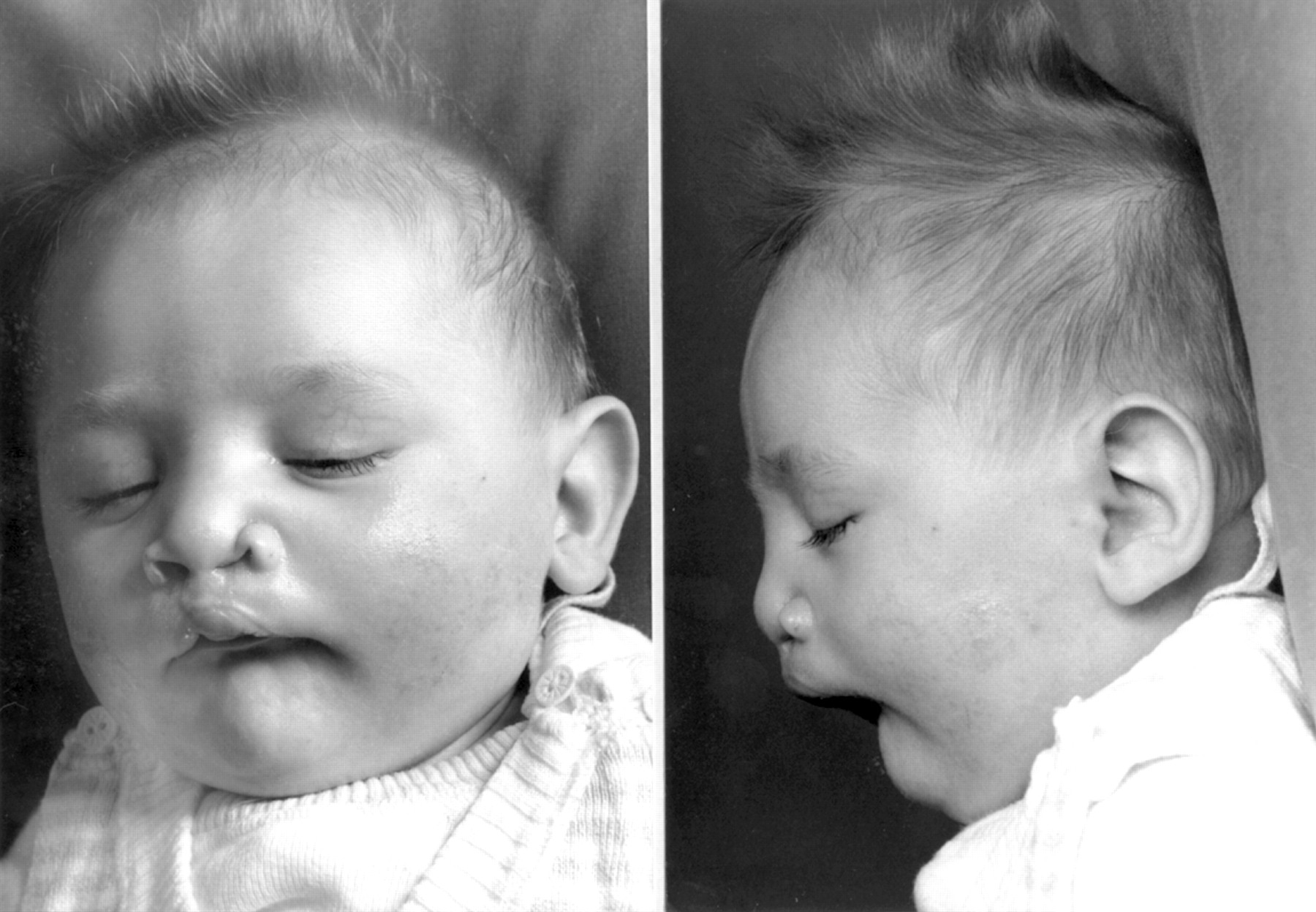
Craniofacial features of the patient aged 3 years. (Photographs reproduced with permission.)
Cytogenetics
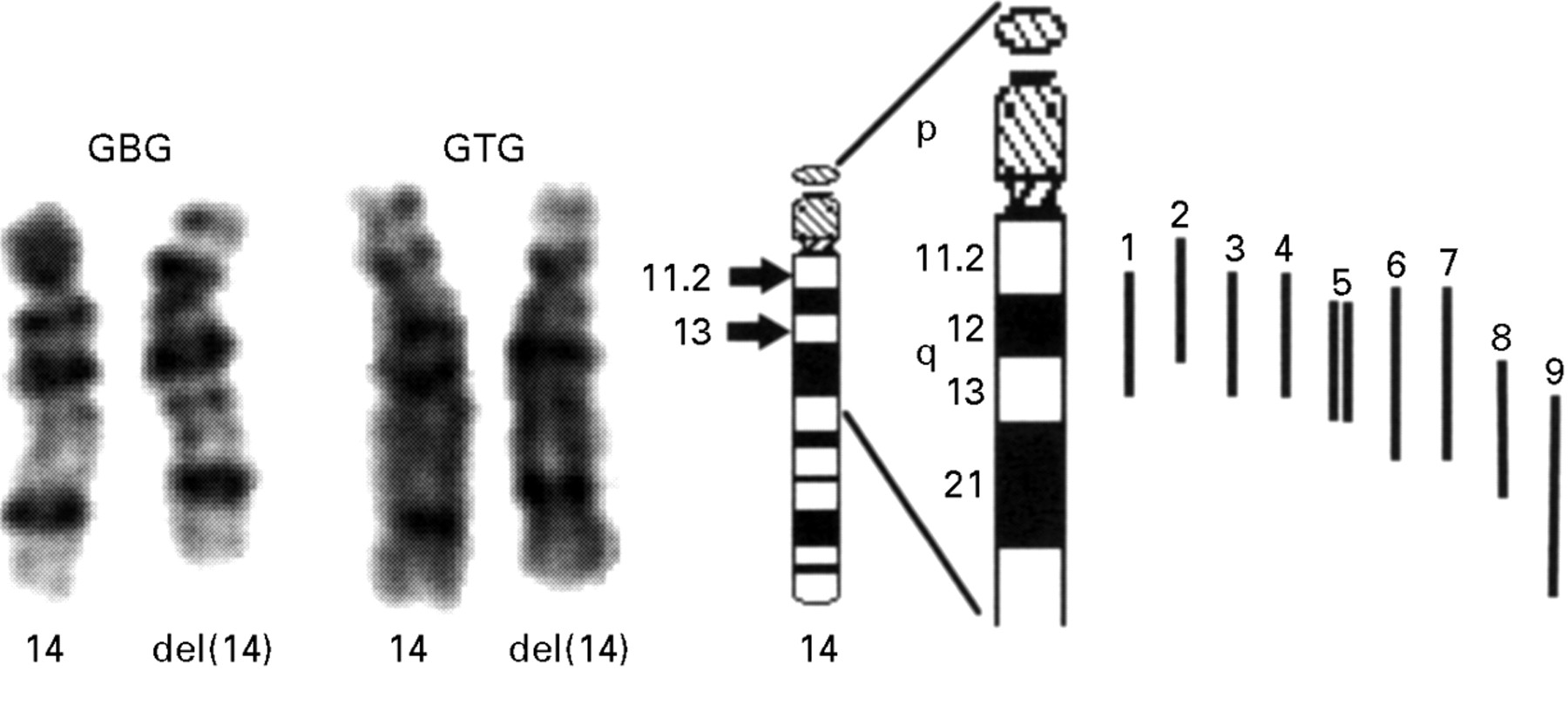
Chromosome analysis of peripheral lymphocytes using standard procedures showed the karyotype 46,XY,del(14)(q11.2q13) (fig 2). Parental karyotypes were normal.

Illustration of the deletion of proximal 14q of the proband and nine other published cases. GBG and GTG stained chromosomes 14 of the proband. The breakpoints are indicated by arrows on the ideogram. Deletions of published cases are indicated as bars in relation to the partial ideogram of chromosome 14. 1: present case, 2: Levin and Surana,6 3: Grammatico et al,2 4: Govaerts et al,1 5: two sisters, Kodoma et al,3 6: case 737, Shapira et al,4 7: Bruyere et al,7 8: Chen et al,5 9: case 777, Shapira et al.4
DNA studies
METHODS
DNA was isolated from EDTA anticoagulated blood of the patient and his parents by standard methods and was typed with six polymorphic (CA)n microsatellite repeats from 14q using the published flanking oligodeoxyribonucleotides as primers.8 Each PCR reaction consisted of 300 ng of genomic DNA, 150 ng of each of the forward and reverse primers, and 10 μCi α32P dCTP. The PCR conditions were as follows: 30 cycles at 94°C for 60 seconds, 55°C for 60 seconds, and 72°C for 90 seconds. The final elongation cycle was seven minutes at 72°C. The amplified products were electrophoresed on a polyacrylamide gel and detected by autoradiography.
For gene dosage analysis of the PAX9 gene, 7 μg of DNA were digested with EcoRI, fractionated on a 0.8% agarose gel, and blotted onto a nylon membrane. The membrane was first hybridised with a 1.1 kb cDNA probe of the PAX9 gene and subsequently with a control probe (IMAGp998I1474), mapped to chromosome 10p.
RESULTS
A total of six polymorphic (CA)n repeats located on proximal 14q were typed (table 1). The affected child had two alleles for D14S72, D14S264, and D14S75 and a single allele inherited from the mother for D14S80 and D14S70, indicating that the deletion was paternally derived. The proximal deletion breakpoint mapped within the 4 cM interval between D14S264 (heterozygous) and D14S80 (hemizygous), and the distal breakpoint mapped within the 4 cM interval between D14S70 (hemizygous) and D14S75 (heterozygous), indicating a deletion size of at least 13 cM. Gene dosage analysis of the PAX9 gene showed hemizygosity in the patient, thus mapping PAX9 within the deletion region (fig 3).
Genotyping of six microsatellite repeat loci in the family and results of deletion analysis of two previously reported patients

Gene dosage analysis of the PAX9 gene. The patient (P) shows reduced dosage of the 6.0 kb EcoRI fragment corresponding to PAX9 (probe 1.1 kb cDNA), while the parents (F, M) and an unrelated control (C) show a normal dosage. The 8.0 kb fragment corresponds to the control probe (IMAGp998I1474) which maps to chromosome 10p.
Fluorescence in situ hybridisation (FISH)
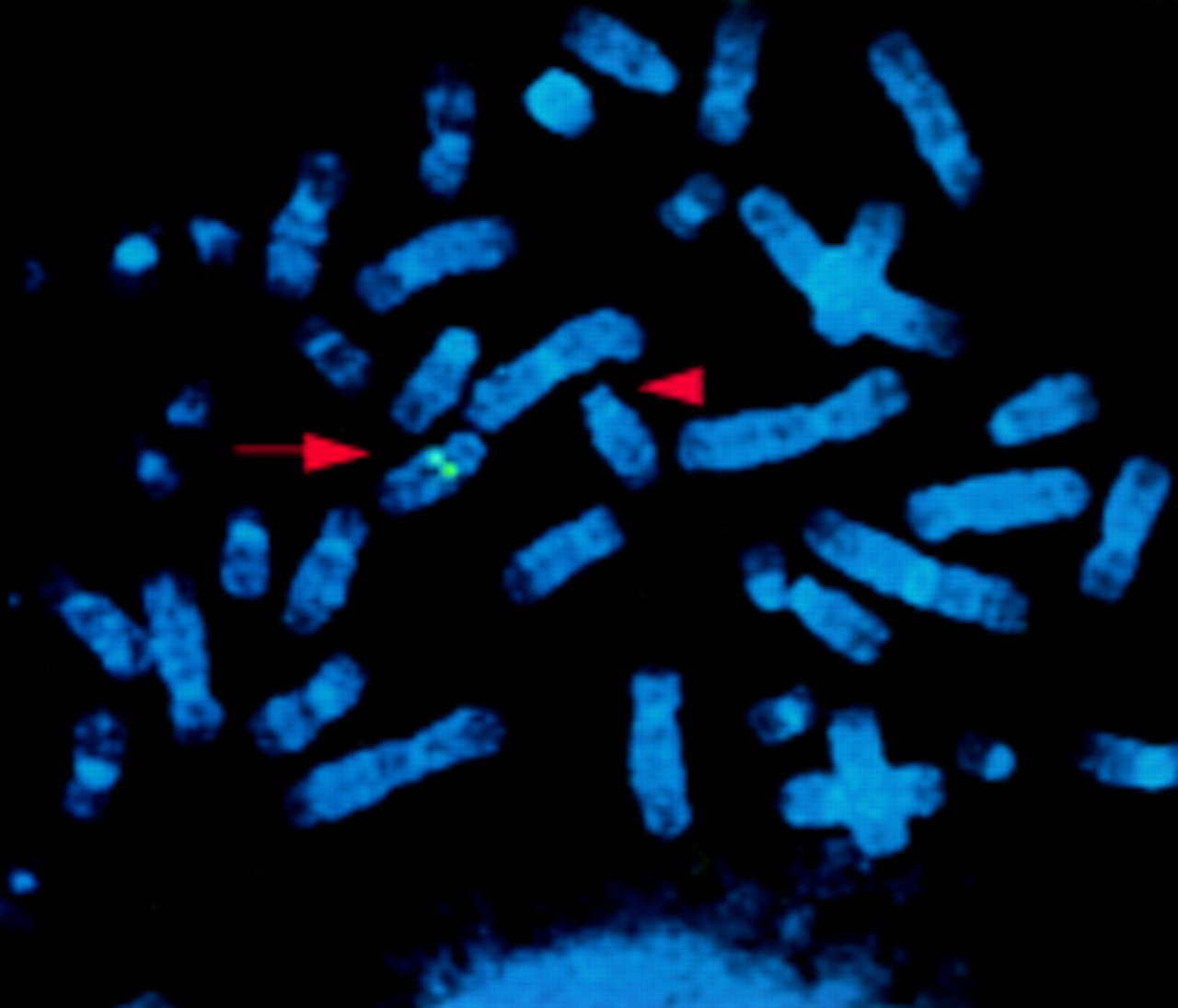
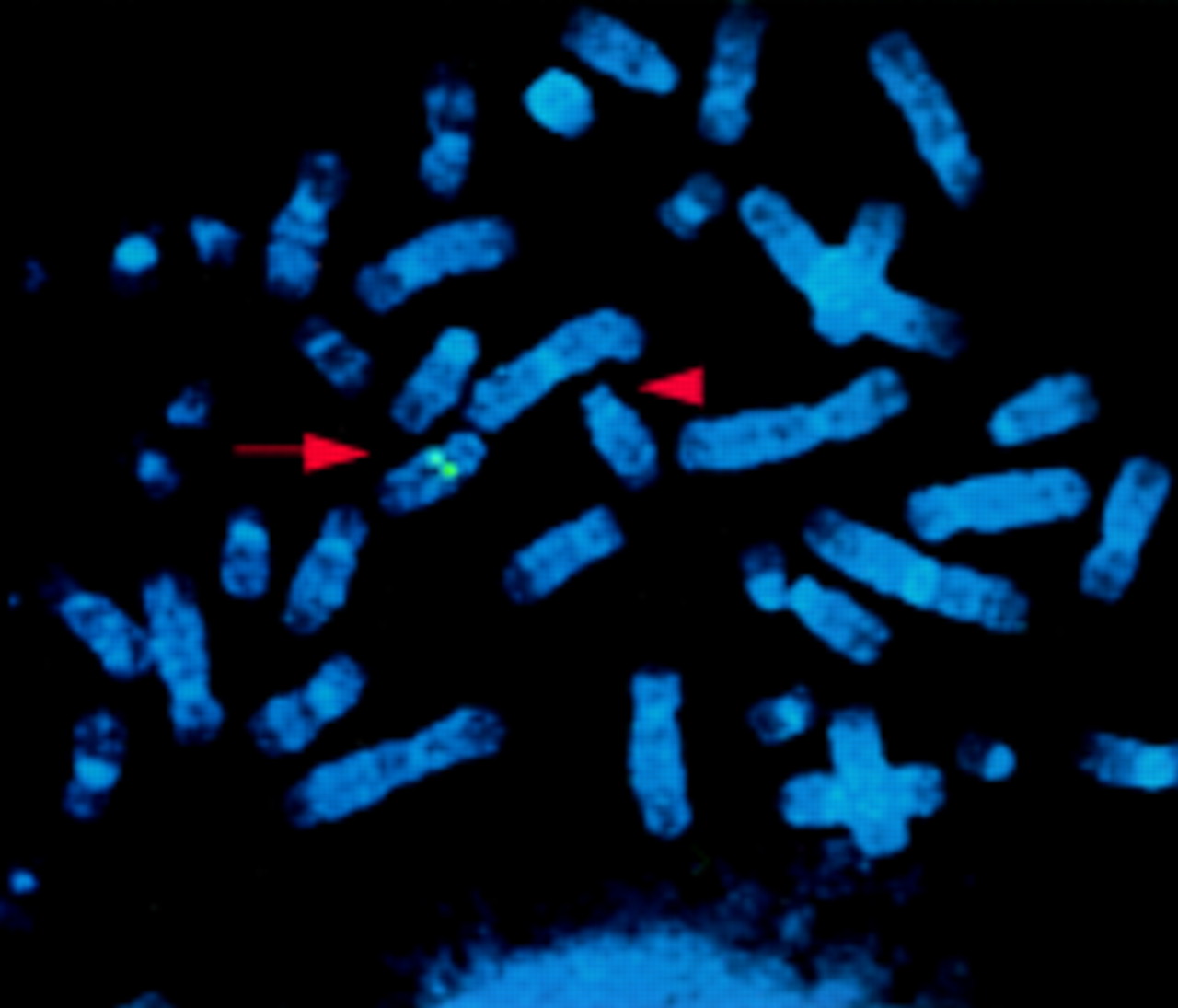
Human PAC clones carrying the PAX9 gene were selected from a human genomic PAC library on filter panels (PAC library RPZIP704, Recource Center/Primary Database, RZPD). The PCR product used as a probe was amplified from human genomic DNA using PAX9 specific primers (PAX9F: 5′-GCT GTG TGT TCA TTT TGC AGA GCC A-3′, PAX9R: 5′-TGG CTC CTG GCA AGA TCG AGC CC-3′) and was 32P labelled by a random primed labelling kit (Boehringer Mannheim). For FISH analysis of metaphase spreads of the proband and of a healthy control, the identified PACs RPCIP704-K12630, -M22554, and -B02531 were labelled by nick translation with biotin-14-dUTP (Gibco, BRL) and preannealed with Cot-1 DNA. Chromosome spreads were obtained from a lymphoblastoid cell line of the patient. Detection and visualisation were performed as described elsewhere9 and chromosomes were identified by staining with 4,6-diamino-2-phenylindolehydrochloride (DAPI). In the control, normal signals were detected on both homologues of chromosome 14 at band q12 (not shown) for all three PACs. The patient showed a signal on the normal chromosome 14 but not on the chromosome 14 with the deletion (fig 4) indicating hemizygosity for PAX9 and confirming the gene dosage analysis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
FISH analysis using PAC RPCIP704-M22554. A normal signal is detected on the normal chromosome 14 at band 14q12 (arrow), but not on the chromosome 14 with the interstitial deletion (arrowhead).
Discussion
PHENOTYPE
Deletions of proximal 14q have been reported in only nine patients to date1-7 (fig 2). The phenotypes of these patients vary substantially. However, a few non-specific symptoms, including growth retardation, muscular hypotonia, psychomotor retardation, and micrognathia, are shared by all these patients and the present case. Our patient was the sole patient with bilateral cleft lip/palate and bilateral colobomas, thereby broadening the phenotypic spectrum associated with partial monosomy of proximal 14q. Moreover, he presented with a severe developmental defect of the brain, resulting in microcephaly, severe muscular hypotonia, cortical blindness, and impairment of temperature regulation. Other congenital anomalies associated with monosomy of proximal 14q involve microcephaly (present case),1 2 4 holoprosencephaly,5-7agenesis/hypoplasia of the corpus callosum (present case),4 patent ductus arteriosus and pulmonary stenosis (case 777),4 arthrogryposis,1 kidney cysts,1 cryptorchidism,2 club feet (case 777),4 and asymmetrical hemisoma.2 This lack of a common pattern of malformations and organ defects might be because of different chromosomal breakpoints (fig 2). The deletion observed in our patient, del(14)(q11.2q13), was one of the most proximal. Interestingly, despite its being cytogenetically identical to those described by Govaerts et al 1and Grammatico et al,2 there was no consistent phenotype in these three patients.
Only two patients, both with more distal deletions, were analysed at a molecular level.4 The deletion in patient 737 overlaps partially with the deletion in our patient, as shown by the hemizygosity of locus D14S70 in both patients (table 1). Hypo-/aplasia of the corpus callosum was the only malformation common to our patient and patient 737.
PARENTAL ORIGIN
Since maternal uniparental disomy of chromosome 14 is associated with short stature and precocious puberty, the existence of one or more imprinted genes on that chromosome is very likely.10 The mapping of such genes might be aided by phenotype analysis in patients with deletions of known parental origin. The deletion (14)(q12q13.3) in two sisters reported by Kodoma et al 3 was maternal in origin, based on cytogenetic 14p polymorphism, suggesting gonadal mosaicism of the clinically unaffected mother. A paternal origin of the deletions of proximal 14q have been determined using cytogenetic polymorphisms of 14p in two patients1 4 and by microsatellite analysis in another two patients (present case, case 7374). These few cases are insufficient to allow correlations of phenotype and parental origin of the deletions.
CANDIDATE GENES
The complex craniofacial malformations in our patient might be caused by haploinsufficiency of one or more genes within the deletion region. Regions flanking the deletion also have to be considered owing to possible position effects. PAX9, a developmental control gene of the PAX gene family, was previously mapped to proximal 14q.11Gene dosage and FISH analysis in our patient showed hemizygosity of PAX9. This confirmed previous mapping data and localised PAX9 within the 21 cM interval between loci D14S264 and D14S75. Our patient is the first report of hemizygosity of PAX9 in humans. During mouse embryogenesis, Pax9 is expressed in the endodermal epithelium lining the pharyngeal pouches, in the vertebral column, and in restricted regions of mesenchyme of the face and limbs.12 Homozygous loss of function ofPax9 causes a variety of malformations in mice including agenesis of the thymus, parathyroid glands, and teeth, cleft palate, and preaxial digit duplication (H Peters, unpublished data). Of these features, only cleft palate was observed in our patient. Hemizygous mice do not show developmental defects (H Peters, unpublished data), possibly arguing against a causal role of PAX9 hemizygosity in the craniofacial malformations of our patient. However, comparison to other PAX genes indicates the difficulties in predicting the phenotypic effect of human PAX mutations on the basis of the findings in the respective mouse mutants. So far, PAX gene mutations in both species are known for PAX2, PAX3, and PAX6. Heterozygous loss of function of PAX6 causes microphthalmia in mice (small eye, sey), but aniridia with a normal eye size in humans.13 14 Mutations of PAX2 are associated with optic nerve coloboma and kidney anomalies in humans.15 The same nucleotide deletion in the murinePax2 gene causes similar eye and kidney abnormalities, but also brain and ear anomalies, not found in humans.16 Mutations of PAX3 resulting in impaired DNA binding capacity cause pigmentary anomalies of the skin in heterozygous mice (splotch, sp) and men (Waardenburg syndrome I and III). However, the hearing loss and inner ear abnormalities commonly found in Waardenburg syndrome are not observed in the mouse mutants. Therefore, normal development of mice hemizygous for PAX9 does not rule out abnormalities caused by PAX9 haploinsufficiency in humans. In addition to organogenesis, PAX9 may also play a role in postnatal life. In adult mice,Pax9 is strongly expressed in the oesophagus and tongue, suggesting a function in the formation and maintenance of the stratified squamous epithelium of the upper digestive tract.17 Interestingly, our patient had dysphagia resulting from reduced oesophageal peristalsis and poor swallowing, which might be related to the reduced PAX9 dosage. Dysphagia is also a symptom of oculopharyngeal muscular dystrophy (OPMD, MIM164300). However, the gene mutated in this disorder, PABP2,18 is flanked by loci D14S990 and D14S1457, which map at least 7 cM proximal to the deletion in our patient.
The NRL gene encoding a leucine zipper transcription factor also maps to proximal 14q.19 In the mouse embryo, it is expressed in postmitotic neuronal cells and developing lens, suggesting a function in eye and neuronal differentiation. Microsatellite analysis in our patient showed heterozygosity for locus D14S64 corresponding to the NRL gene. Exclusion of NRL from the deletion makes a causal role of NRL in the bilateral coloboma of our patient unlikely.
Based on the deletion size of at least 13 cM, our patient is expected to be hemizygous for more than 100 genes. Many of them may play a role in embryonic development, such as the transcription factor genes forkhead-like 1 and 2 (FKHL1, FKHL2), which have been mapped to 14q1320 21 and are very likely to be hemizygous in our patient. To prove haploinsufficiency of such candidate genes, molecular analysis of further cases with partial monosomy of proximal 14q and studies on mouse models are necessary.
Acknowledgments
We thank K B Jedele and T Meitinger for critical reading of the manuscript and I Krägeloh-Mann for critical review of the neurological data. This study was supported by the Deutsche Forschungsgemeinschaft.