Article Text

Abstract
Background The structural organisation of the human IGF2/ICR1/H19 11p15 domain is very complex, and the mechanisms underlying its regulation are poorly understood. The Imprinted Center Region 1 (ICR1) contains seven binding sites for the zinc-finger protein CTCF (CBS: CTCF Binding Sites); three additional differentially methylated regions (DMR) are located at the H19 promoter (H19DMR) and two in the IGF2 gene (DMR0 and DMR2), respectively. Loss of imprinting at the IGF2/ICR1/H19 domain results in two growth disorders with opposite phenotypes: Beckwith–Wiedemann syndrome and Russell Silver syndrome (RSS). Despite the IGF2/ICR1/H19 locus being widely studied, the extent of hypomethylation across the domain remains not yet addressed in patients with RSS.
Methods We assessed a detailed investigation of the methylation status of the 11p15 ICR1 CBS1-7, IGF2DMR0 and H19DMR (H19 promoter) in a population of controls (n=50) and RSS carrying (n=104) or not (n=65) carrying a hypomethylation at the 11p15 ICR1 region.
Results The methylation indexes (MI) were balanced at all regions in the control population and patients with RSS without any as yet identified molecular anomaly. Interestingly, patients with RSS with ICR1 hypomethylation showed uneven profiles of methylation among the CBSs and DMRs. Furthermore, normal MIs at CBS1 and CBS7 were identified in 9% of patients.
Conclusions The hypomethylation does not spread equally throughout the IGF2/ICR1/H19 locus, and some loci could have normal MI, which may lead to underdiagnosis of patients with RSS with ICR1 hypomethylation. The uneven pattern of methylation suggests that some CBSs may play different roles in the tridimensional chromosomal looping regulation of this locus.
- Imprinting Disorders
- 11p15 region
- Russell Silver Syndrome
- IGF2/H19 domain
- Growth retardation
Statistics from Altmetric.com
Introduction
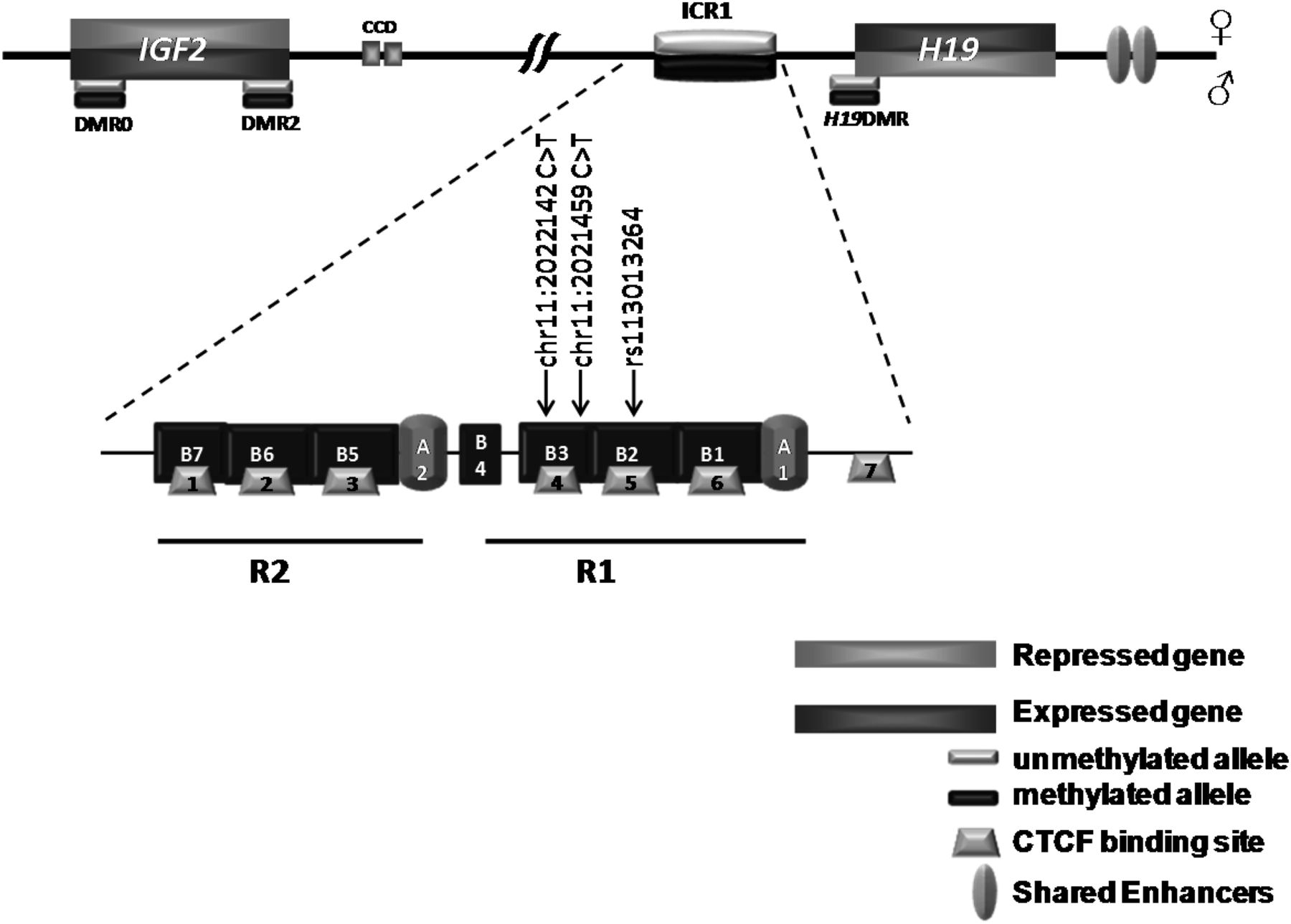
The IGF2/ICR1/H19 11p15 locus mapping to the human telomeric chromosome 11p15.5 region is one of the imprinted loci playing an important role in fetal and postnatal development and tumorigenesis. This cluster is regulated by its own imprinting control region, ICR1.1 IGF2 and H19 share a common set of tissue-specific enhancers located downstream of H19.2–5 An additional intergenic-enhancer called Centrally Conserved Domain (CCD) is located between the ICR1 and IGF2.6 ,7 While the CCD regulates biallelic expression of Igf2 in plexus/choroid tissues in mice,8 its role remains unknown in humans. The ICR1, located 2 kb upstream of the H19 promoter, which is methylated on the paternal allele during spermatogenesis (germline differentially methylated regions (DMR)), controls the monoallelic expression of IGF2 and H19 by insulating communication between the H19 downstream enhancers and the IGF2 and H19 promoters. The chromatin insulator property of the IGF2/ICR1/H19 locus is regulated by a chromatin insulator protein, CCCTC binding factor (CTCF) which binds only the unmethylated maternal allele where it prevents the shared enhancers to activate IGF2 promoters but activates the H19 promoter. By contrast, on the paternal allele, the ICR1 methylation prevents CTCF binding and allows the shared enhancers to access IGF2 promoters leading to its expression.9 Figure 1 describes the organisation of the IGF2/ICR1/H19 locus with the main regions involved in its imprinting regulation.

Schematic representation of the 11p15 IGF2/ICR1/H19 domain showing the different differentially methylated regions (DMR). The lower panel represents a zoom of the ICR1 organisation. The positions of the SNP affecting CBS5 TaqMan probes, as well as those of the newly identified variations, are depicted. CCD, centrally conserved domain; ICR, imprinting control region; R, repeat; CBS, CTCF binding sites.
The human ICR1 consists of two subdomains (R1 and R2), each of which contains a repeated sequence A, followed by three or four repeats B. Each sequence B (with the exception of B4 which is incomplete) has a binding site for CTCF (CTCF Binding Sites (CBS)). ICR1 contains seven CBSs (CBS 1–6 are contained in R1 and R2, and CBS7 is outside of them).10 ,11 In addition to ICR1, the IGF2/ICR1/H19 11p15 telomeric domain harbours several other DMRs. There are two DMRs in IGF2 (DMR0 and DMR2) and an additional DMR located at the H19 promoter (H19DMR); all of them are somatic DMRs methylated postzygotically on the paternal allele (secondary DMR) (figure 1).12
Loss of imprinting at the 11p15 ICR1 locus is involved in two fetal and postnatal growth disorders, Russell Silver syndrome (RSS: OMIM #180860) and Beckwith–Wiedemann syndrome (BWS: OMIM #130650),13–16 with RSS leading to fetal growth restriction and BWS to fetal overgrowth. More than 50% of patients with RSS display a hypomethylation at 11p15 ICR1.15 This abnormality leads to the downregulation of IGF2 and the biallelic expression of H19. Conversely, 10% of patients with BWS have a hypermethylation at the ICR1, leading to IGF2 overexpression and the downregulation of H19.
The regulation of the human IGF2/ICR1/H19 11p15 locus is complex and poorly understood. Some valuable insights regarding the regulation of this locus have come from the study of patients with RSS or BWS who were found to harbour genetic defects (microdeletions, cis-duplications and point mutations). Indeed, it has been shown that CTCF function is modulated by neighbouring DNA-binding factors, such as the cohesin (involved in the long-range looping at this locus) and pluripotency factors OCT4/SOX2.17 Mutations and small deletions of OCT4-binding and SOX2-binding sites have been described within the ICR1 in patients with BWS, and are associated with a gain of ICR1 methylation.18–20 These results suggest that OCT4/SOX2 bind to the maternal allele and protect it from gain of methylation during the early stages of fetal development.21 ,22
Molecular diagnosis of RSS and BWS is usually made on a few sites of the 11p15 ICR1 locus using different technologies. In our laboratory, we performed the molecular diagnosis of RSS and BWS using two tests; one on the ICR1 CBS2 and the other on the H19DMR (H19 promoter) with a recently developed quantitative real-time PCR (TaqMan allele-specific methylated multiplex real-time quantitative PCR (ASMM RTQ-PCR)).23–25 In this study, we asked whether (1) the hypomethylation occurred homogeneously at the IGF2/ICR1/H19 domain in patients with RSS carrying ICR1 CBS2 hypomethylation and (2) abnormal methylation at the IGF2/ICR1/H19 11p15 domain could be identified in RSS without 11p15 ICR1 CBS2 hypomethylation. For this purpose, we analysed, in detail, the extent of the methylation pattern across the IGF2/ICR1/H19 locus in a large cohort comprising controls, RSS with ICR1 CBS2 hypomethylation and RSS without a yet identified anomaly using ASMM RTQ-PCR and pyrosequencing.
Material and methods
Population study
The study was conducted in accordance with national ethics regulations (agreement numbers 681 and 682, Assistance Publique-Hôpitaux de Paris). Informed consent for genetic analysis was obtained from patients or their parents. Genomic DNA was extracted from leucocytes of each control and patient using standard techniques.
Controls
The control population consisted of adults and children, all healthy individuals (n=50).26 ,27
Patients
One hundred and sixty-nine patients with RSS were analysed. Of these, 104 patients were diagnosed with RSS with ICR1 CBS2 hypomethylation (10 of them were published elsewhere27), and 65 patients with RSS with no identifiable molecular anomaly (neither ICR1 CBS2 11p15 hypomethylation nor maternal UPD of chromosome 7).
Bisulfite treatment
Genomic DNA (1 µg) was sodium bisulfite treated with an EZ DNA methylation kit (Zymo Research, Orange, California, USA), according to the manufacturer's instructions, and then eluted in 50 µL RNase-free H2O.
TaqMan ASMM RTQ-PCR
The ASMM RTQ-PCR, a TaqMan assay for the analysis of the seven ICR1 CBSs, IGF2DMR0, IGF2DMR2, H19DMR (H19 promoter) (GenBank AF125183) and 11p15 ICR2 (GenBank: NW 001838018.2), was performed as described previously23 ,24 using 7900HT Fast Real-Time PCR System (Life Technologies, France). Briefly, the multiplex amplification was performed in 96-well plates in a reaction volume of 20 µL containing 10 ng of sodium bisulfite-treated genomic DNA (3 µL per well), 10 µL TaqMan Master Mix, 900 nM of each primer (Sigma Aldrich, France) and 200 nM of each TaqMan-MGB probe (Life Technologies, France). The analysis has been performed using SDS V.2.4 software (Life Technologies, France). The methylation index (MI) at each locus has been assigned by calculating the ratio between the methylated and unmethylated alleles as follows: (amount of methylated allele/sum of both methylated and unmethylated alleles) × 100. For each locus, we have considered an MI as normal if it was within the range expressed, with mean MI value of the control population ±2SDs and abnormal when outside of this range. TaqMan Master Mixes and amplification conditions, as well as primer and probe sequences, are available upon request.
DNA methylation analysis by pyrosequencing
Quantitative DNA methylation analysis was performed by pyrosequencing of bisulfite-treated DNA.28 One microgram of DNA was bisulfite converted using the EpiTect 96 Bisulfite kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Regions of interest for validation were amplified using 30 ng of bisulfite-treated human genomic DNA and 5–7.5 pmol of forward and reverse primer, one of them being biotinylated. Sequences for oligonucleotides for PCR amplification and pyrosequencing are available upon request. Reaction conditions were 1× HotStar Taq buffer supplemented with 1.6 mM MgCl2, 100 µM dNTPs and 2.0 U HotStar Taq polymerase (Qiagen, Courtaboeuf, France) in a 25 µL volume. The PCR programme consisted of a denaturing step of 15 min at 95°C followed by 50 cycles of 30 s at 95°C, 30 s at the respective annealing temperature, and 20 s at 72°C, with a final extension of 5 min at 72°C. Ten microlitres of PCR product was rendered single-stranded, as previously described,28 and 4 pmol of the respective sequencing primer was used for analysis. Quantitative DNA methylation analysis was carried out on a PSQ 96MD system with the PyroGold SQA Reagent Kit (Qiagen Courtaboeuf, France), and the results were analysed using the PyroMark CpG software (V.1.0.11.14, Qiagen Courtaboeuf, France).
ICR1 and OCT4/SOX2 motifs sequencing
ICR1 (including OCT4/SOX2 motif two (OSM2) in A2 repeat and OSM3 in B3) and OSM1, lying upstream of ICR1, were sequenced as previously described26 by standard methods using the ABI PRISM Big Dye Terminator V.3.0 Cycle Sequencing Kit, and an ABI 3100 Genetic Analyzer. The sequencing products were then analysed with SeqScape V.2.6 (Life technologies, Courtaboeuf, France).
Statistical analysis
Principal component analysis (PCA) was performed on data to identify patterns at the IGF2/ICR1/H19 11p15 loci.
Agreement between ASMM RTQ-PCR and pyrosequencing, in terms of qualitative results, was assessed by calculating the κ coefficient.29 Intraclass correlation coefficients and κ coefficients indicate the degree of similarity between the results obtained with the two methods. An ICC or κ coefficient greater than or equal to 0.9 was considered excellent.
Results
The molecular diagnosis of RSS is routinely established in our laboratory according to the methylation status of ICR1 CBS2 and H19DMR (H19 promoter) using ASMM RTQ-PCR. Here we studied the methylation status of the IGF2/ICR1/H19 11p15 imprinted domain at all regulatory elements (IGF2DMR0, IGF2DMR2, the seven CBSs at ICR1 and H19DMR) in leukocytes, DNA extracted from a collection of patients presenting with RSS (104 RSS with hypomethylation at 11p15 ICR1 CBS2 and H19DMR and 65 RSS with no identified molecular defect) and control subjects (see online supplementary figure S1). We used quantitative methods for DNA methylation analysis (ASMM RTQ-PCR and pyrosequencing) to investigate the regions of interest and define the extent of epigenetic abnormalities. Furthermore, we sequenced the entire ICR1 seeking for genetic abnormalities that might affect cis-regulatory elements.
Control subjects
To establish the range of normal MIs, we first analysed the methylation at different loci of the IGF2/ICR1/H19 domain of DNA from the control population, using both ASMM RTQ-PCR (figure 2A) and pyrosequencing (figure 2B,C). The MIs calculated for the control subjects were normally distributed, which allowed us to calculate a mean MI and SD. The intra-assay and interassay coefficients were calculated using ASMM RTQ-PCR for each locus (see online supplementary table S1). Two MIs were calculated for data obtained by the pyrosequencing method: one from all CpGs included in the amplification product and sequenced by pyrosequencing (MICBS-pyro); the second from only those CpGs interrogated by ASMM RTQ-PCR (MICpG-pyro). We have further compared MIs between ASMM RTQ-PCR (MIASMM) and pyrosequencing. We considered an MI to be normal if it was within two SDs of the mean MI value for control subjects and abnormal if it was outside this range.

Methylation indexes of the IGF2/ICR1/H19 11p15 domain obtained by either ASMM RTQ-PCR (A) or pyrosequencing (B, C) in control subjects. MICBS-Pyro refers to the MI calculated from the sum MIs of each CpG amplified and sequenced by pyrosequencing. MICpG-Pyro refers to the MI calculated from the sum of MIs of each CpG covered by the TaqMan probe used in the ASMM RTQ-PCR and analysed by pyrosequencing. Each circle represents an individual MI. The bars represent mean±1SD. MI, methylation index; CBS, CTCF binding sites; DMR, differentially methylated regions; ASMM RTQ-PCR, TaqMan allele-specific methylated multiplex real-time quantitative PCR.
MICBS-pyro and MICpG-pyro calculated by pyrosequencing were comparable (see online supplementary table S2). The comparison between the two methods revealed that MIASMM for the IGF2DMR2 and ICR1 CBS5 were more variable than those obtained by pyrosequencing. The pyrosequencing study allowed documenting the methylation status of all the CpGs included in the sequence of interest and the identification of SNPs. Pyrosequencing revealed that the CpG7 of the IGF2DMR2 (included in the TaqMan probes used to study IGF2DMR2 by ASMM RTQ-PCR) was more often hypermethylated in controls (CpG7 mean MI=67±7.3; n=24) compared with the neighbouring CpGs (see online supplementary table S2). The variability of ICR1 CBS5 MIASMM could be attributed to the SNP rs113013264 located in the hybridisation sites of the ASMM RTQ-PCR TaqMan probe (figure 1 and online supplementary table S2). These two loci were excluded from further analysis.
Patients with RSS with no identified molecular defect
We analysed 65 patients diagnosed with RSS according to our scoring system.15 The 11p15 ICR1 CBS2, as well as H19DMR MIs, was normal for all the patients (see online supplementary figure S1 and figure 3). Furthermore, we searched for abnormal methylation at other ICR1 CBSs or in the IGF2DMR0 that could explain the phenotype of these patients. All the MIASMM for ICR1 CBS1–4, 6–7, H19DMR and IGF2DMR0 were in the normal ranges (figure 3). These results exclude a methylation defect in the IGF2/ICR1/H19 domain as a locus involved in the aetiology of these patients.
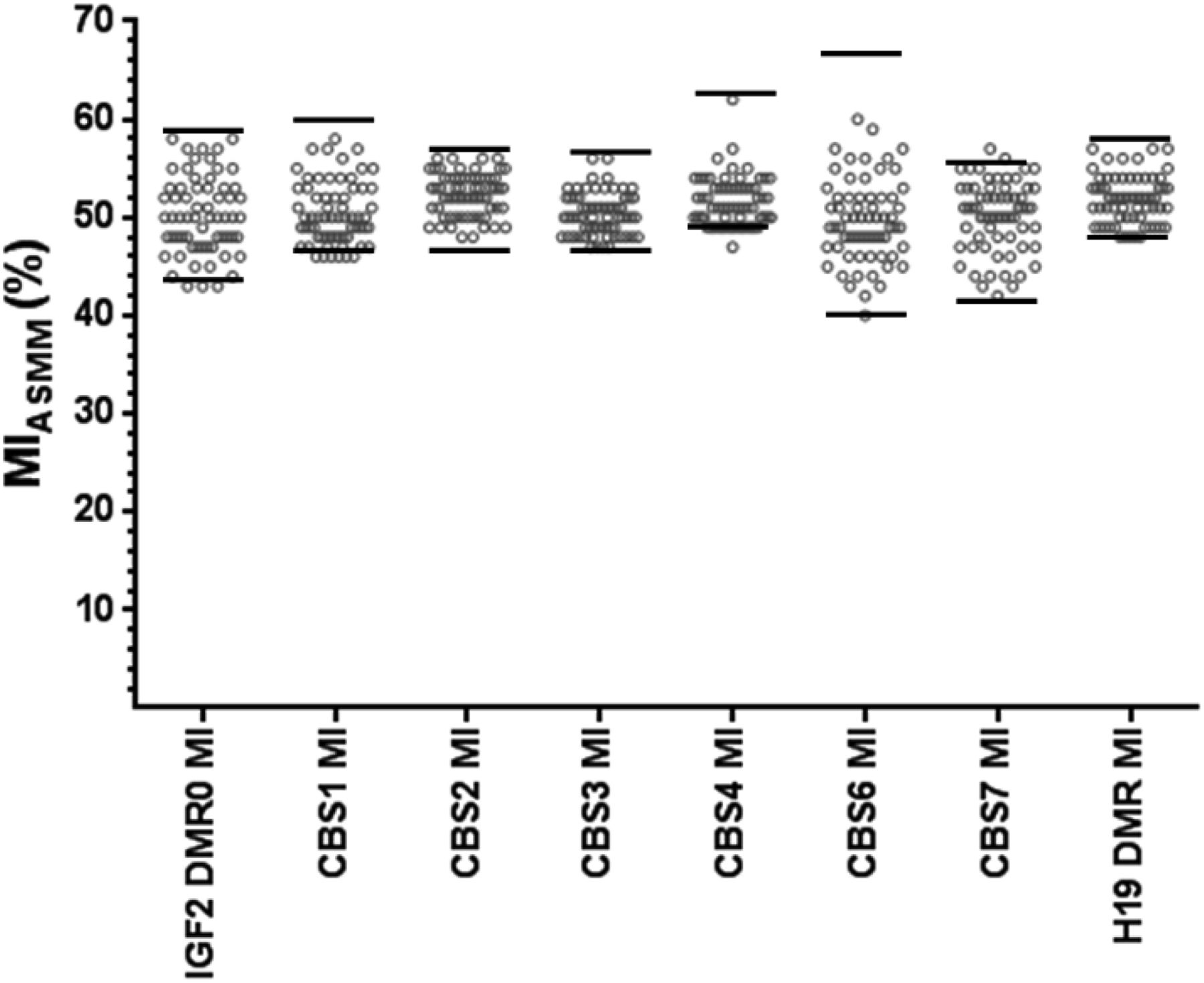
Methylation indexes of the IGF2/ICR1/H19 domain obtained by ASMM RTQ-PCR for patients with Russell Silver syndrome without identified anomaly. Each circle represents an individual MI. The two horizontal bars circumscribe the range of normal MI values calculated from the control population (mean±2SD). MI, methylation index; CBS, CTCF binding sites; DMR, differentially methylated regions; ASMM RTQ-PCR, TaqMan allele-specific methylated multiplex real-time quantitative PCR.
Distribution of methylation abnormalities along the IGF2/ICR1/H19 11p15 domain in patients with RSS with hypomethylation at the ICR1 CBS2
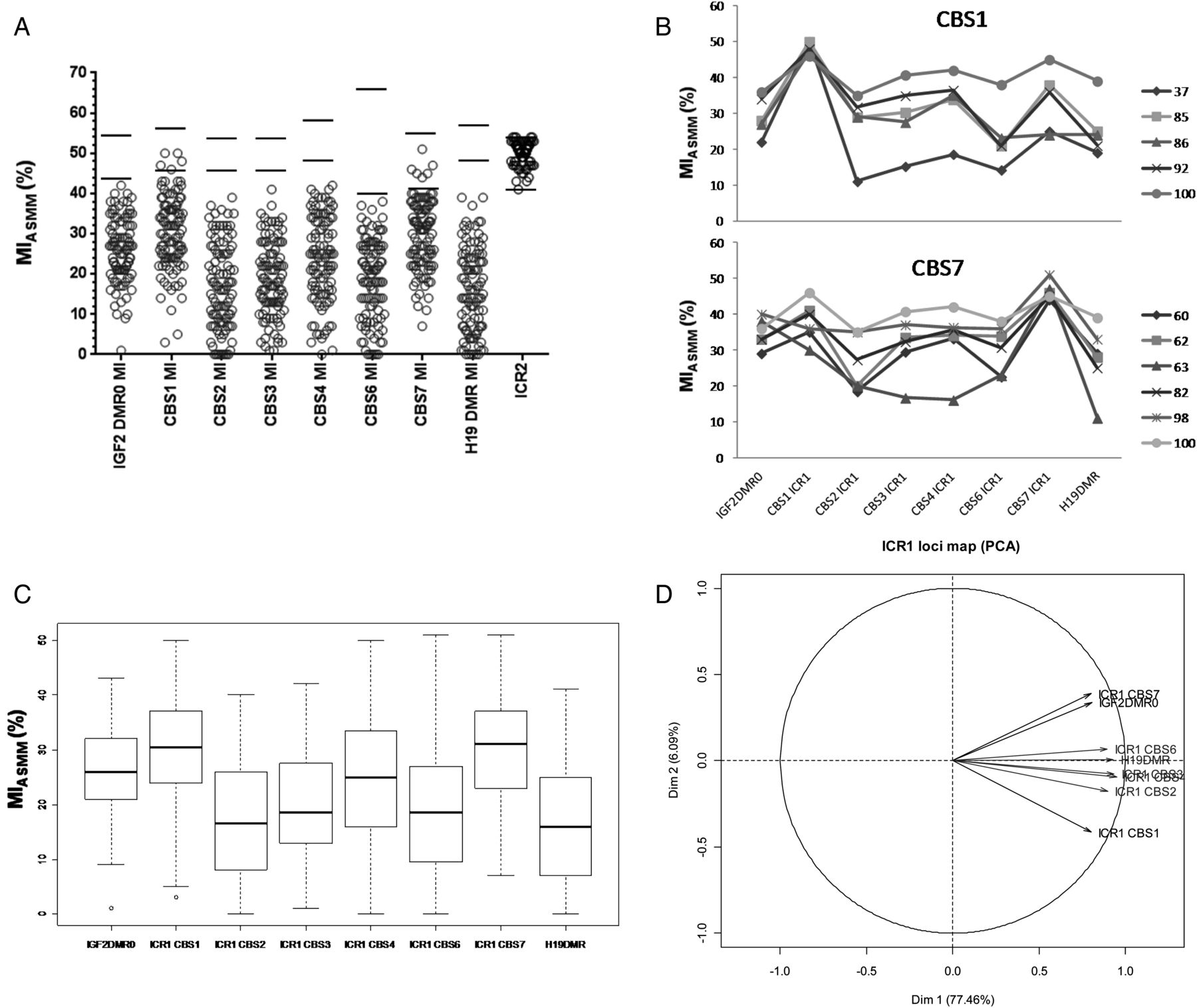
We then sought to investigate the spatial extent of the methylation abnormality in the IGF2/ICR1/H19 domain. We, therefore, studied 104 patients with RSS exhibiting hypomethylation at 11p15 ICR1 CBS2 and H19DMR. The methylation status at 11p15 ICR2 was assessed as an internal control. All patients with RSS carrying ICR1 CBS2 hypomethylation showed normal methylation level at the 11p15 ICR2 locus.
Almost all patients exhibited hypomethylation at all the IGF2/ICR1/H19 11p15 domain loci analysed. Interestingly, five and six of the 104 patients with RSS (9% overall) showed normal MIASMM either at the ICR1 CBS1 or CBS7, respectively (figure 4A,B). Only one patient (RSS100) showed hypomethylation at both ICR1 CBS1 and CBS7 (figure 4B). Furthermore, the hypomethylation was more pronounced at ICR1 CBS2, CBS3, CBS4 and CBS6 and H19DMR, which were found to be highly correlated (table 1 and online supplementary table S4) compared with the ICR1 CBS1, CBS7 and IGF2DMR0 loci (figure 4A, C and online supplementary table S4). For instance, when ICR1 CBS2 MI is <20%, large numbers of patients have similar levels of hypomethylation at ICR1 CBS3 (81%), ICR1 CBS4 (56%), ICR1 CBS6 (75%), and H19DMR (81%). Oppositely, only 32%, 21% and 24% of patients were similarly hypomethylated at IGF2DMR0, ICR1 CBS1 and ICR1 CBS7, respectively, as at ICR1 CBS2 (see online supplementary table S4).
Pearson tests showing the correlation between IGF2/ICR1/H19 11p15 loci
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Methylation indexes of the IGF2/ICR1/H19 11p15 domain obtained by ASMM RTQ-PCR for patients with Russell Silver syndrome carrying ICR1 CBS2 hypomethylation. Each circle represents an individual MI. (B) Pattern of MIs across the IGF2/ICR1/H19 11p15 obtained for the patients carrying normal MIs either at ICR1 CBS1 (upper panel) or ICR1 CBS7 (lower panel). (C) Box plot of methylation index in ICR1 region. (D) Principal components analysis (correlation of variables to the axis) showing how the different CBSs covariate together. ICR1 CBS2, CBS3, CBS4 and CBS6 and H19DMR are hypomethylated to the same extent and form the major axis (77.76%) of the variability. The second minor axis is formed by ICR1 CBS1, CBS7 and IGF2DMR0 with particularity ICR1 CBS7 and IGF2DMR0 which are more methylated than ICR1 CBS1. The two horizontal bars circumscribe the range of normal MI values calculated from the control population (mean±2SD). MI, methylation index; CBS, CTCF binding sites; DMR, differentially methylated regions; ASMM RTQ-PCR, TaqMan allele-specific methylated multiplex real-time quantitative PCR; PCA, principal component analysis.
In order to better understand the correlation between these loci, we performed a PCA on the calculated MIs. This analysis showed that the largest proportion of variability is summarised by the first axis of the PCA (77.76%), with a significant contribution of ICR1 CBS2, CBS3, CBS4 and CBS6 and H19DMR (figure 4D). The patients with RSS are generally hypomethylated to the same extent at these loci. By contrast, ICR1 CBS1, CBS7 and IGF2DMR0, although related, were found to be less hypomethylated compared with the former group. The second axis of the analysis showed that the patients were also differentially hypomethylated at ICR1 CSB1, CBS7 and IGF2DMR0. This axis opposed patients with higher MIs at the ICR1 CBS7 and IGF2DMR0 compared with the ICR1 CBS1 (figure 4D).
To draw detailed comparisons, and to confirm these results, we sequenced the same regulatory region using pyrosequencing for 34 patients with RSS. Agreement between the two tests, in terms of MIs concordance, was assessed by calculating the κ coefficient.29 A κ coefficient of ≥0.9 was considered excellent. Table 2 shows that the κ coefficients obtained varied from good to excellent for all loci. When considering the qualitative results for the patients with RSS (ie, hypomethyaltion or not), the κ coefficient equals 1, which indicates a perfect agreement between the two methods (data not shown).
Coefficient agreement, κ, representing the concordance between MIASMM and MIpyrosequencing
MIs calculated from the two technologies were concordant for ICR1 CBS1 when comparing MIASMM and MICpG-pyro or MICBS-pyro. Similarly, we sequenced five out of the six patients with RSS with ICR1 CBS7 normal MIASMM and found concordant MIs with pyrosequencing (see online supplementary table S3).
ICR1 and OCT4/SOX2-binding sites sequencing
Genetic abnormalities have been reported in patients with either BWS or RSS.30 Furthermore, we recently reported OCT4/SOX2-biding site mutations in a subset of patients with BWS with a mismatch ICR1 hypermethylation between ICR1 CBS2 and H19DMR MI.26 We searched for cis-genetic abnormalities in the 11 patients showing normal MI at ICR1 CBS1 or ICR1 CBS7. For this purpose, we sequenced the entire ICR1 locus (4.5 kb) including OSM2 and OSM3, as well as OSM1 upstream of ICR1. We identified two de novo heterozygous variations (chr11:2021459 C>T in RSS37 carrying normal MI at CBS1 and chr11:2022142 C>T in RSS82 carrying normal MI at CBS7; online supplementary table S3); the first one is upstream and outside of CBS4, and the second one was within CBS4 but did not affect the CTCF consensus binding site (figure 1). These variations were neither found in control subjects (100 chromosomes) nor in the databases. The analysis of the MIs across the IGF2/ICR1/H19 of these patients did not reveal any evident pattern that would suggest a putative molecular mechanism involving these two variations (figure 4B).
Discussion
The human IGF2/ICR1/H19 11p15 domain is a complex locus harbouring several regions, all methylated on the paternal allele and all involved in the regulation of the domain presumably in different ways. Although it is widely studied, the fine-tuning regulation of this domain is still poorly understood in humans. In a large proportion of patients with RSS, a hypomethylation of the 11p15 ICR1 can be identified.15 However, the laboratory tests are developed to interrogate only particular sites and do not give an exhaustive landscape of the abnormality across the entire domain. In this study, we investigated genomic DNA methylation at 10 loci of the IGF2/ICR1/H19 11p15 domain in a large cohort of patients with RSS displaying 11p15 ICR1 CBS2 and H19DMR hypomethylation to give more insight regarding the molecular basis of the regulation of this complex domain.
We first analysed 65 patients with RSS without any as yet identified molecular cause, in an attempt to identify methylation abnormalities within regions other than the ICR1 CBS2 and H19DMR that we investigated routinely for the molecular diagnosis of RSS. All MIs calculated for this cohort were within the normal range, which exclude, thereby, a methylation anomaly of the IGF2/ICR1/H19 11p15 domain to be involved in the phenotype of these patients. These data suggest that other candidate regions, yet to be identified, might cause RSS. Indeed, the molecular abnormalities causing RSS remain unknown in about 30% of cases.15 Recently, some rare genetic abnormalities, mainly copy number variations, have been identified in patients with an RSS phenotype, but these are far from representing all molecular causes of RSS31 ,32 (and brought out in this article).
Interestingly, the methylation analysis of the IGF2/ICR1/H19 domain in RSS carrying ICR1 CBS2 and H19DMR hypomethylation revealed that this abnormality did not spread equally over the entire domain, but rather showed an uneven pattern of methylation. Indeed, we observed that the hypomethylation found at the ICR1 CBS2, CBS3, CBS4 CBS6 and H19DMR was more frequently pronounced than that occurring at the other loci analysed (IGF2DMR0, ICR1 CBS1, CBS7); which may suggest that the former loci are mutually coregulated. Furthermore, 9% of patients showed normal MI at ICR1 CBS1 or ICR1 CBS7, located at the boundaries of the ICR1 domain. Kannenberg et al33 showed similar results on five patients with RSS. However, in that study, the authors investigated only five CpGs located in the H19DMR and, therefore, did not give exhaustive information on the entire IGF2/ICR1/H19 domain as did our study. More recently, we have identified similar results in a survey of a BWS cohort carrying ICR1 11p15 hypermethylation.26 Indeed, we have found that a subset of patients with BWS may have normal MIs at IGF2DMR0, CBS1, CBS4, CBS6 or CBS7. By contrast with BWS, we have identified normal MI only at CBS1 and CBS7 in patients with RSS. This suggests that different mechanisms operate to protect the unmethylated maternal alleles from gaining methylation, or the methylated allele from losing methylation.
It has been demonstrated in mice, that the methylation of the Igf2/ICR1/H19 loci did not spread linearly but is, instead, established in a hierarchical manner under the control of ICR1. These data suggest the involvement of long-range chromatin interactions in this process.34 CTCF plays a critical role in the long-range chromatin interaction at the IGF2/ICR1/H19 domain. Recent studies have shown that CTCF function is modulated by neighbouring DNA-binding factors, such as the cohesin and pluripotency factors OCT4/SOX2.17 The higher-order chromatin looping occurs in a different manner on the two parental chromosomes and is mediated by CTCF-cohesin binding and correlates with DNA methylation and histone modifications. Furthermore, this higher-order chromatin looping is abolished in RSS or BWS cell lines with ICR1 11p15 methylation anomalies.35 ,36 However, even though these studies demonstrate that DNA methylation is crucial for proper long-range interaction at the IGF2/ICR1/H19 domain, they do not give an explanation for the uneven pattern of hypomethylation seen in our study.
In light of all those studies, it appears that the most likely hypothesis that might explain our results is the involvement of a cis-acting or trans-acting factor that protects the paternal allele from the DNA hypomethylation during early stages of embryogenesis. This hypothesis is supported by recent findings demonstrating the importance of OCT4/SOX2 binding in the protection of the maternal allele from hypermethylation at Igf2/ICR1/H19 domain in blastocysts.21 ,22 ,37 We have identified two novel unreported de novo variations in two patients, but they are unlikely to be the cause of the hypomethylation since they do not disturb a known cis-acting element and also they did not induce any particular methylation profile across the domain. In humans, a subset of maternally inherited mutations affecting OCT4/SOX2 binding sites at A2 repeat have recently been reported in a patient with BWS with 11p15 ICR1 hypermethylation.18–20 ,26 Interestingly, these abnormalities induced more pronounced hypermethylation around the mutations, and the degree of hypermethylation decreased when moving away from the site of the mutations.22 ,26 ,37 Likewise, in this study, we showed that the ICR1 middle part (CBS2–CBS6), as well as H19DMR, is hypomethylated at a similar level in patients with RSS. These findings suggest that a similar mechanism, yet to be identified, as that operating to protect the maternal allele from hypermethylation, may operate to prevent the paternal allele from hypomethylation.
In conclusion, taking together the fact that the methylation spreads hierarchically over the IGF2/ICR1/H19 domain, the domain undergoes long-range chromatin interactions mediated by CTCF and other cofactors (cohesin, OCT4/SOX2…) and the involvement of transfactors in the protection against hypermethylation or hypomethylation during critical periods of embryogenesis, the hypothetical mechanism explaining our results would be as follows: ICR1 CBS2, CBS3, CBS4, CBS6 and H19DMR are engaged together in the chromatin interactions within the domain, while IGF2DMR0, ICR1 CBS1 and CBS7 are not, and might be located inside the loops or excluded from them. These interactions could be mediated by a yet unknown transfactor. An abnormality in that trans-acting factor or its binding site on the domain will prevent the protection of the paternal allele from demethylation which would be more pronounced on the first group of loci than the other, and ICR1 CBS1 and CBS7 located at extremities of ICR1 could be spared from the demethylation wave crossing the domain.
Acknowledgments
We thank the patients, their families and the physicians for referring patients and collecting clinical data. We thank Dr Anne Segonds-Pichon (Bioinformatics Department, Babraham Institute, Cambridge CB22 3AT, UK) for helping us with the statistical analysis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Contributors SA designed the study and wrote the paper. VS, AB and WAH performed the experiments. JT and FB performed the pyrosequencing experiments and analysed the data; JT revised the manuscript. NT, CDN and MLJ performed RSS molecular diagnosis at 11p15 in leucocytes DNA. YK performed the statistical analysis. SR analysed the data and revised the manuscript. YLB and IN designed and supervised the study and revised the manuscript.
Funding This work was supported by INSERM, UPMC-Paris6 funding, ANR EPIFEGRO 2010, Pfizer grant, Agence de Biomédecine 2010 grant. WAH was supported from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007-2013/ under REA grant agreement n° 290123. SA was supported by Novonordisk and the INSERM-ANR EPIFEGRO 2010. IN is member of the COST Action BM1208.
Competing interests None.
Ethics approval Assistance Publique-Hôpitaux de Paris.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Correction