Article Text

Abstract
The identification of somatic driver mutations in cancer has enabled therapeutic advances by identifying drug targets critical to disease causation. However, such genomic discoveries in oncology have not translated into advances for non-cancerous disease since point mutations in a single cell would be unlikely to cause non-malignant disease. An exception to this would occur if the mutation happened early enough in development to be present in a large percentage of a tissue's cellular population. We sought to identify the existence of somatic mutations occurring early in human development by ascertaining base-pair mutations present in one of a pair of monozygotic twins, but absent from the other and assessing evidence for mosaicism. To do so, we genome-wide genotyped 66 apparently healthy monozygotic adult twins at 506 786 high-quality single nucleotide polymorphisms (SNPs) in white blood cells. Discrepant SNPs were verified by Sanger sequencing and a selected subset was tested for mosaicism by targeted high-depth next-generation sequencing (20 000-fold coverage) as a surrogate marker of timing of the mutation. Two de novo somatic mutations were unequivocally confirmed to be present in white blood cells, resulting in a frequency of 1.2×10−7 mutations per nucleotide. There was little evidence of mosaicism on high-depth next-generation sequencing, suggesting that these mutations occurred early in embryonic development. These findings provide direct evidence that early somatic point mutations do occur and can lead to differences in genomes between otherwise identical twins, suggesting a considerable burden of somatic mutations among the trillions of mitoses that occur over the human lifespan.
Keywords: somatic mutation; monozygotic twins; genome-wide genotyping; next-generation sequencing; genetic discordance
- somatic mutation
- monozygotic twins
- genome-wide genotyping
- next-generation sequencing
- genetic discordance
Statistics from Altmetric.com
- somatic mutation
- monozygotic twins
- genome-wide genotyping
- next-generation sequencing
- genetic discordance
Introduction
Mutation is an important source of genetic variation in the human genome. It can introduce deleterious nucleotide changes to genes or provide fuel for phenotypic evolution. It is well accepted that such mutations may lead to cancer,1 but it is unlikely that all base-pair mutations would cause malignancy. Rather, such stochastic events could also lead to possible disruption of an organ's function, particularly so if the mutation were present in a large proportion of the cells within a tissue. Such mutations could exist if they were introduced early in embryogenesis and their identification could identify important control points in disease aetiology, such as has happened for malignant disease.
Indeed, somatic mutations have previously been demonstrated to cause non-malignant disease. Rapid advances in molecular genetics have demonstrated the importance of somatic mutation in a great variety of human diseases other than cancer (reviewed elsewhere).2 For instance, a recent proof-of-concept study has revealed the existence of early embryonic somatic mutations causing Dravet syndrome,3 as well as another novel finding of mosaic AKT1 mutation in a Proteus syndrome patient.4 A more recent publication identified somatic mutations in individuals with clonal haematopoiesis but without haematopoietic malignancies.5
In addition, the identification of early somatic mutations would provide preliminary insights into somatic mutation rates, which have been previously estimated in in vitro cell models6–10 or disease–gene data.11 ,12 Recent advances in sequencing technology have provided such rates in tumour samples,13–16 and genotyping arrays have led to the detection of copy number variation in population studies.17–19 However, the burden of point mutations in non-malignant tissue is not known, and further we are not aware of previous reports identifying the existence of somatic point mutations in otherwise apparently healthy individuals.
Monozygotic twins provide a natural experiment to address these questions since any differences between monozygotic co-twins would arise due to somatic changes. In the current study, we genome-wide genotyped 66 monozygotic twins (33 pairs) to identify somatic point mutations. In addition, we sought validation of the candidate somatic mutations through Sanger sequencing. Given that mutations occurring later in organ development would lead to somatic mosaicism within a cell line, we assessed the degree of mosaicism of identified mutations using next-generation sequencing. Together, these data provide direct evidence of the existence of somatic mutations occurring early in human development and a preliminary estimate of the rate at which they occur in an otherwise normal population.
Results
Data generation
By genome-wide genotyping with Illumina 610K single nucleotide polymorphism (SNP) arrays, in the same laboratory at the same time, we obtained genotype calls on 506 821 SNPs in 33 pairs of monozygotic twins (66 individuals) after quality control (figure 1). We only included high-quality (per SNP missing rate <0.01, Hardy–Weinberg Equilibrium p>10−6) common SNPs (minor allele frequency >0.05). The cut-off of genotyping call rate per sample was 5%.

Flowchart of study design. MZ, monozygotic; HWE, Hardy–Weinberg Equilibrium; MAF, minor allele frequency. Access the article online to view this figure in colour.
The call rate in the 66 individuals included in this study was >99.9%. Zygosity of the twins was determined by standardised questionnaire and confirmed by genotype concordance on a genome-wide level (>99.9%) in the 33 pairs of twins. To identify potential somatic mutations, we first assessed the genome-wide genotypes and identified 1087 discordant SNPs across all twin pairs. Of these, 1019 were present once across all twin pairs, 64 were present in two pairs of twins and 4 SNPs were discordant in three pairs of twins. Excluding these four SNPs as a potential source of genotyping error (since some SNPs are more difficult to genotype than others), we observed a median of 10 discordant SNPs per pair of monozygotic twins ranging from 1 to 282 (mean=34.76).
Raw data inspection to identify candidate somatic mutations for external verification
Despite precautions to select only the highest quality SNPs, the majority of the discordant SNPs were likely to be due to genotyping errors. To resolve this possible source of error, we visually inspected the cluster plots of the 1087 discordant SNPs between 66 monozygotic twins (33 pairs). Examination of the data (cluster separation, tightness, intensity and outsider genotype) suggested that 93% of these discordances most probably represented base-calling errors. Next we selected only SNPs that were called as being in the centre of a fluorescent cluster on the genotyping cluster plots, so as to preferentially identify mutations that were not mosaic, since mosaic mutations would more likely be between two clusters rather than at the centre of a cluster. We identified 80 SNPs as potentially early somatic mutations and carried out validation experiments on all these SNPs.
Verification by Sanger sequencing
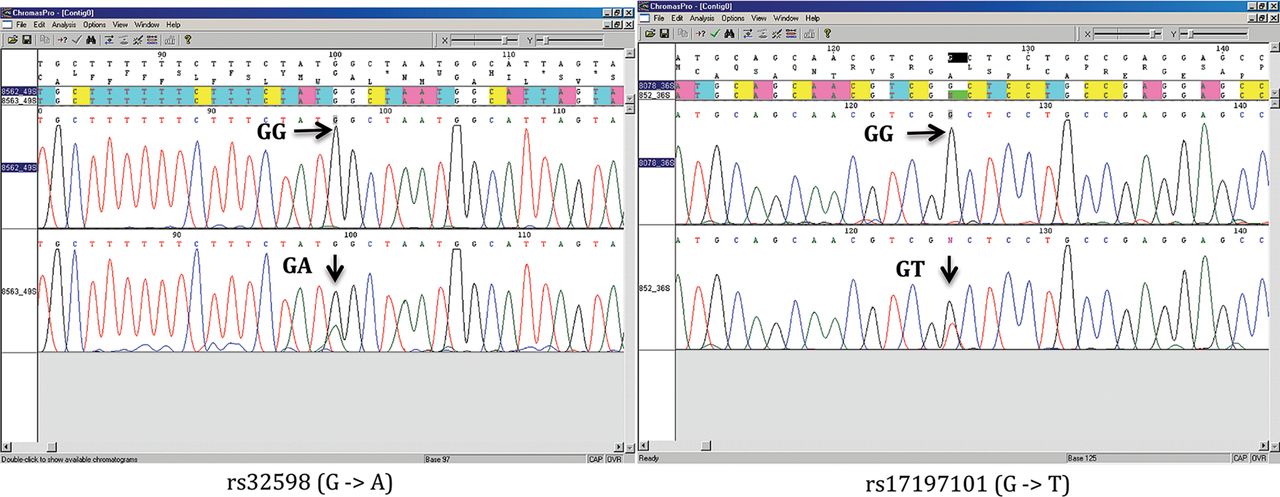
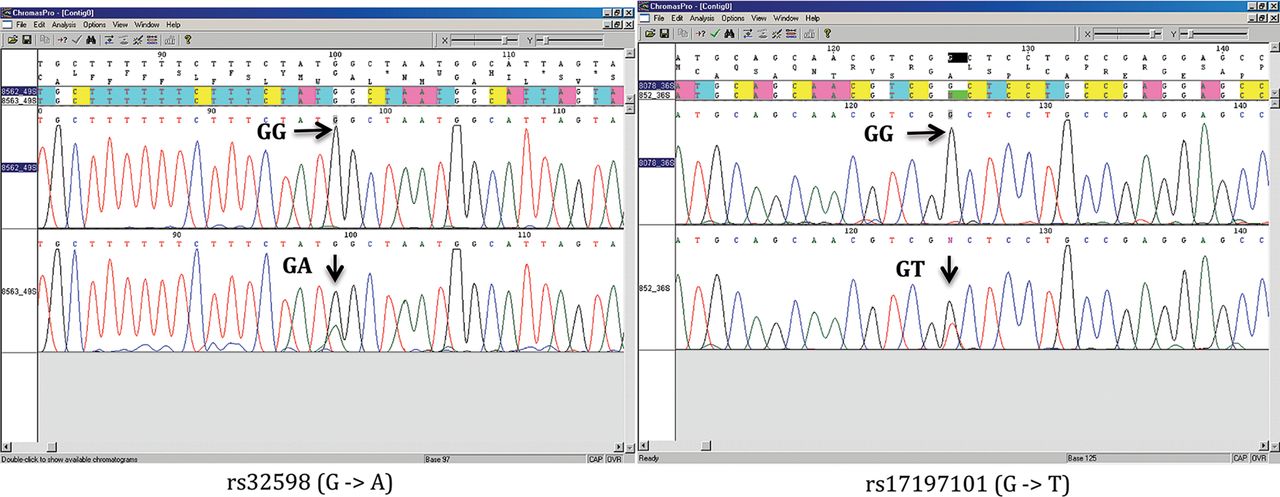
We next amplified the region spanning each candidate somatic mutation from each pair of genotype discordant monozygotic twins and sequenced them by conventional Sanger sequencing for 80 discordant SNPs in 33 monozygotic twins from initial data set. Only 2 out of 80 (2.5%) candidate somatic mutations were confirmed from two distinct pairs, with one twin being homozygous and the other twin being heterozygous (figure 2). This low verification rate suggested that few additional mutations would be identified if we relaxed the quality control criteria in the analysis on genome-wide genotyping data.
{kind=link}
{kind=link}
Sanger sequencing chromatography of identified somatic mutations. Access the article online to view this figure in colour.
Mutation validation and somatic mosaicism exploration by next-generation sequencing
Somatic mutations occurring at a late stage in the exponential cellular expansion phase of development would be only present in a small proportion of cells and would result in somatic mosaicism.20 ,21 It has been suggested that Sanger sequencing has limited sensitivity to detect such mosaic somatic mutations,22–24 and therefore, in order to interrogate mutations for mosaicism, we further applied high-depth next-generation sequencing as a complementary approach for 18 SNPs that showed evidence of possible somatic mosaicism in Sanger sequencing and the two somatic mutations verified by Sanger sequencing as positive controls. Equal amounts of PCR–amplicon DNA from 20 different genomic regions in 20 distinct twin pairs were pooled in two samples while DNA from co-twins was placed in separate pools randomly. Using the Illumina HiSeq, we generated 23 Gb of sequence for each of the pooled samples with an average of ∼20 000-fold coverage per amplified region (see online supplementary table S1).
Aligning the sequencing reads to the human reference genome (build 37), the two somatic mutations identified by Sanger sequencing were confirmed. The reference allele over all alleles ratio (RAF), as determined by allelic reads, was 1 for the homozygous twin while the RAF was 0.5 and 0.6 for each of the heterozygous twin, respectively (table 1). The reads counted prior to PCR duplicates removal gave the same ratio as that subjected to duplicates removal. We did not identify additional evidence for somatic mutations from the 18 other candidate mutant single nucleotide variants (SNVs) (see online supplementary table S1).
Somatic mutations verified by Sanger sequencing and next-generation sequencing
Preliminary measurement of somatic mutation frequency
The total number of sites investigated was determined from the intersection of high-quality SNPs called in both co-twins that met the quality control criteria as shown in online supplementary table S2. The somatic mutation frequency was calculated as the number of mutation events divided by the sum of total number of SNP sites in each twin pair. The estimated mutation frequency was 1.2×10−7 per base pair.
We observed little evidence for striking mosaicism from the next-generation sequencing results based on RAF (see online supplementary table S1). Since we observed an RAF of 0.5–0.6 for the heterozygous twin at mutant sites, it is highly probable that the mutations we observed were generated at the cell division when two monozygotic twins were separated, which results in the almost 50:50 ratio of two alleles if the mutation causing one reference allele become the alternative allele (see online supplementary figure S1). Given that the mutation may occur in either co-twin, we estimate the somatic mutation rate as 6.0×10−8 per nucleotide per cell division. We note that this estimate is preliminary and is discussed below.
Given that the human genome is estimated to contain approximately 3.0×109 bp25 (human genome build 37 V.2: 3156 Mb), it is likely that each individual carries approximately over 300 postzygotic mutations in the nuclear genome of white blood cells that occurred in the early development.
Mutations observed
Of the two somatic mutations we observed, one is a G to A transition (rs32598) located on chromosome 5q14, which is an intronic SNP in the EGF-like repeats and discoidin I-like domains 3 gene (EDIL3, [MIM XXX 606018]). The other mutation is represented by a transversion from G to T (rs17197101) in a CpG dinucleotide site located on chromosome 6p21.33. This SNP resides in 5′UTR of the Transcription factor 19 gene (TCF19, [MIM 600912]). In the Catalogue Of Somatic Mutations In Cancer (COSMIC),26 multiple types of somatic mutations, including nonsense substitutions and missense substitutions, have been found for EDIL3 in cancer cells, and two mutations have been found for TCF19. In spite of this, no mutations have been found in haematopoietic or lymphoid cancers for these genes. Thus, it is unlikely that either change conferred a proliferative or survival advantage that would interfere with the above calculations.
Discussion
In this study, we have identified and verified somatic point mutations occurring in otherwise healthy individuals early in development. Our preliminary estimates of the rate of such mutations suggest that this phenomenon occurs relatively frequently. Whether such mutations may rarely give rise to non-malignant disease requires further investigation, but such exploration may identify important physiologic control points in disease aetiology rendering novel drug targets, much like the identification of driver mutations in cancer has led to important therapeutic advances in oncology.
Driver mutations in cancer have highlighted critical drug targets whose manipulation has led to changes in disease survival. For example, somatic mutations in vascular endothelial growth factor (VEGF) leading to non-small cell lung cancer27 respond to bevacizumab, which neutralises VEGF.28 And likewise somatic mutations in ERBB2 (also called HER2) are present in lung and breast cancers,29 and successful targeting of this tyrosine kinase with trastuzumab improves outcomes.30 Despite these advances in malignant disease, point mutations as a cause of non-malignant disease have received little attention since such mutations would only rarely underlie disease causation and they would need to happen early enough in development, or confer a non-malignant proliferative advantage, to lead to organ disruption and consequent disease. However, despite their rarity, their identification would render a drug target that would be sufficient to cause disease.
Despite these limitations, there is evidence that somatic mutations can lead to rare diseases. McCune–Albright syndrome is due to mosaicism for an activating somatic mutation.31 Somatic spontaneous reversions of disease-causing mutations to the wildtype allele occur in Fanconi anaemia32 and other autosomal recessive disorders.33–36 Thus, somatic mutations can rarely cause non-malignant disease.
Monozygotic twins are considered an attractive model for studying somatic variations. Previous studies identified large structural variants such as different copy number profiles17 ,37 and chromosomal aneuploidies.38 Genetic differences in monozygotic twins have also been demonstrated in epigenetic marks39 and DNA changes.3 However, in most cases, the somatic variations were observed in a later stage of life17 ,39or related to disease phenotype,3 leaving the burden of single-point mutation arising in early life poorly described. Our study surveyed the magnitude of these kind of mutations across the entire genome. Using normal adult monozygotic twins, we provide evidence of relatively frequent mutations originating in early development.
Most early studies on somatic mutation rate only captured the ones causing impaired function in coding region or non-coding regulatory region,6 ,11 ,12 which excludes the majority of the genome. In addition, several other factors could have an effect on mutation estimate derived from gene-based studies, such as gene structure and sensitivity of mutation detection methods. Accounting for all these factors, the average somatic point mutation rate has been estimated as 7.7×10−10 per nucleotide per cell division,40 which is much lower than our estimate (6.0×10−8). Moreover, a recent study sequencing whole genomes of a CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) trio detected a somatic mutation frequency of 2.52×10−7 per nucleotide41 by distinguishing germline and somatic mutation through validation from a third generation of the same family. Although this estimate is two times ours (1.2×10−7), it is nevertheless influenced by unavoidable additional mutations arising in cell lines.
Our measure of mutation rate has a number of limitations, which will be overcome by future studies. First, the uncertainty of mutation rate could be improved substantially by more accurate cell depth estimate using advanced technology, for instance, a well-established method estimating cell depth based on somatic mutations in microsatellites.42 Second, we only assessed common SNPs, which may not fully represent the nature of mutation at sites of very low alternate allele frequencies.43 Large-scale sequencing projects on monozygotic twins, for example, the UK10K project, will be able to assess the mutation rate at rare SNPs, but will always be encumbered by the relatively high error rates of next-generation sequencing. Third, comparing other cell types of different lineages than leucocytes would better assess the timing of these somatic mutations. However, only blood samples were collected from these subjects and both twin pairs did not permit sampling of other tissues.
Our data provide evidence that early somatic mutations do occur, and based on the mutation frequency observed in our study, it can be inferred that each individual carries, on the average, greater than ∼300 postzygotic mutations that occurred early enough to be present in the majority of blood cells. These mutations are not constrained by the need to be compatible with whole-organism survival when they first occur. However, they provide fuel for phenotypic variation, which may lead to tissue disruption and disease in later life. It has been speculated that, for any gene, a small number of individuals in the population carry a somatic mutation in a large majority of cells that have occurred in early development.20 Our data provide evidence to support this concept.
In summary, our findings support the emerging consensus that somatic point mutations arising in development are a relatively common phenomenon. The mutation frequency we observed may serve as the first step in refining important parameters influencing somatic non-malignant disease.
Materials and methods
Data generation
The monozygotic twins for this study were collected from 3512 individuals in the TwinsUK cohort that have undergone a genome-wide scan using DNA sample extracted from blood. The TwinsUK cohort includes approximately 12 000 volunteer twins from all over the UK,44 who have no differences in age-matched characteristics compared with the general population.45 We performed the genome-wide genotyping by using the Infinium 610k assay (Illumina, San Diego, USA) at two facilities: the Center for Inherited Diseases Research (CIDR) in Baltimore, Maryland, USA, and the Wellcome Trust Sanger Institute in UK. Co-twins that were not genotyped in the same centre at the same time were excluded to avoid the possible sources of spurious mutation findings caused by lab-based systematic differences in genotyping. We used the Illuminus calling algorithm46 to assign SNP genotypes. Null genotypes were assigned if an individual's most likely genotype was called with less than a posterior probability threshold of 0.95. Only the highest quality common SNPs (minor allele frequency >0.05) that had a call rate greater than 0.99 were retained for analysis. In addition, we filtered SNPs demonstrating deviation from Hardy–Weinberg Equilibrium (p<10−6). The zygosity of the twins was confirmed based on concordance rates. SNP genotypes were compared between monozygotic co-twins to identify putative somatic mutations for the following analysis. We obtained informed consent from all participants before they entered the studies. Ethics approval was obtained from the local research ethics committee.
Data filtering to identify candidate somatic mutations for verification
We trimmed the list of putative early somatic mutations via visual inspection of intensity data. The normalisation and base calling were done using Illumina Genome Studio Genotyping Module V.1.9.4. SNPs with discordant calls between co-twins were manually reviewed.
Verification by Sanger sequencing
For the filtered candidate somatic mutations, we performed PCR amplification using 30 ng of genomic DNA from each of the twins. We designed PCR primers by using Primer-3 (http://frodo.wi.mit.edu/) to amplify PCR products in size from 260 to 572 bp (supplementary note). We used High Fidelity Taq Polymerase (error rate is 4×10−6) from Roche in PCR. PCR products were sent for Sanger sequencing and also used to form two pools for next-generation sequencing. Each twin from each pair was randomly assigned to one of the two pools, A or B.
Mutation verification and somatic mosaicism exploration by next-generation sequencing
Before pooling, PCR products were quantified by the PicoGreen kit (Life Technologies Canada, Burlington ON) and 160 ng of each PCR fragment was added to the pool. The pooled DNA was then seared in a Covaris S200 sonicator at peak incident power set at 175 and 200 cycles per burst for 430 s to obtain a fragment distribution with a peak around 150 bp (see online supplementary figure S2). Each pool was separately barcoded. Libraries were constructed by following Illumina pair-end library protocol. The resulting library for each pool was sequenced in a single lane of an Illumina HiSeq GAII analyser flow cell to generate 101 bp paired-end reads according to the manufacturer's instructions.
Sequencing data analysis and somatic mutation identification
For Sanger sequencing, somatic mutations were identified by directly comparing genotypes of co-twins from chromatograms. For next-generation sequencing, the paired-end reads were aligned to reference genome (build 37) using Burrows-Wheeler Aligner (BWA, V.0.6.1-r104) with standard parameters.47 Further postalignment steps were undertaken to minimise false-positive findings from error-prone sequencing results: (1) SAMtools (V.0.1.18-r982:295)48 was used to synchronise the mate pair information. Alignments were filtered for mapping and pairing. (2) PCR duplicates were removed by Picard tools (V.1.77) (http://picard.sourceforge.net/). (3) Genome Analysis Toolkit (GATK, V.2.0)49 was used to realign the reads on the edges of potential insertions and/or deletions to reduce SNV artefacts. (4) Recalibration was also applied to base quality scores by using GATK. SAMtools was used to identify single-nucleotide variations and obtain allelic depth. Allelic depth was used to generate RAF. RAF was given by NR/(NA+NR), in which NR and NA are the number of reads containing reference allele and alternative allele, respectively.
Calculations of somatic mutation frequency
The somatic mutation frequency, f, is calculated as the number of mutation events (m) divided by the sum of total number of SNP sites (S) in each twin pair (T):
All 33 twin pairs were considered in the calculation. The total number of sites investigated was determined from the intersection of high-quality SNPs called in both co-twins that met the quality control criteria as shown in online supplementary table S2. We note that somatic mutations could possibly occur in the hybridisation probe; however, genotypes were generated for SNP sites on the Illumina assay. Thus, only the total number of SNPs assayed (not including those on the probe) was considered for calculation of mutation frequency.
We estimated the mutation rate by the following formula6:
where μ is the single-nucleotide mutation rate, 2 is used to quantify the fact that the mutation may occur in either co-twin, f is the frequency of somatic mutations confirmed by Sanger sequencing experiments and n is the number of cell division that had occurred up to the time of the mutation.
Our study was only able to detect mutations occurring in the exponential growth phase of development rather than tissue renewal20 since we employed genome-wide genotyping for mutation identification. Since the RAF was approximately 0.5 and 1 in heterozygous and homozygous twins, respectively, such a mutation most likely has occurred at the cell division when two twins were separated. Thus, n was estimated to be 1.
Acknowledgments
We would like to thank Vince Forgetta, Zari Dastani and Kelly Burkett for their insightful comments.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online note
- Data supplement 3 - Online table 1
- Data supplement 4 - Online table 2
Footnotes
-
Contributors JBR, CP, MP and RL conceived the study. TS provided the original data. RL, BR, CP and CMTG developed statistical methodologies. RL and AM cleaned and analysed the data. MR, SYMW and CP generated validation data. RL, BR, CP and AM drafted the manuscript. All authors revised the final manuscript. BR is the guarantor.
-
Funding This work was partially supported by Québec Consortium for Drug Discovery.
-
Competing interests None.
-
Ethics approval Ethics approval was obtained from the local research ethics committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.