Article Text

Abstract
Background Heterogeneous molecular defects affecting the 11p15.5 imprinted gene cluster are associated with the opposite growth disorders Beckwith-Wiedemann Syndrome (BWS) and Silver Russell syndrome (SRS). Maternal deletions of the centromeric domain usually result in BWS, but paternal deletions have been so far associated with normal phenotype. Here we describe a case of recurrent severe Intra-Uterine Growth Restriction (IUGR) with paternal transmission of an 11p15.5 60 kb deletion.
Methods and results Chromosome microarray (CMA), PCR and DNA sequencing analyses showed that two fetuses conceived by a normal couple inherited from their father a 60 kb deletion encompassing the Imprinting Control Region of the 11p15.5 centromeric domain. The two fetuses died in utero with severe growth restriction. PCR amplification of parental DNAs indicated that the father carried the mutation in the mosaic state. DNA methylation and gene expression analyses showed that the deletion led to an imprinting alteration restricted to the centromeric domain and resulting in silencing of KCNQ1OT1 and activation of CDKN1C and PHLDA2.
Conclusions Our data demonstrate that the phenotype associated with 11p15.5 deletions is strongly influenced by the size of the region involved and indicate imprinting defects leading to CDKN1C and PHLDA2 activation as cause of severe IUGR.
- Epigenetics
- Imprinting
- Chromosomal
- Molecular genetics
Statistics from Altmetric.com
The monoallelic and gamete of origin-dependent expression of imprinted genes requires the presence of specific imprinting control regions (ICRs).1 These are sequences characterised by differential DNA methylation on the maternally and paternally derived chromosomes. A large cluster of imprinted genes resides at chromosome 11p15.5, and is organised in two regulatory domains, each including a specific ICR. The telomeric domain includes the IGF2 and H19 genes. Opposite epigenetic and genetic defects have been associated with the overgrowth disorder, Beckwith–Wiedemann Syndrome (BWS, OMIM 130650),2 and the undergrowth disorder, Silver–Russell Syndrome (SRS, OMIM 180860).3 The larger centromeric domain includes several growth-related genes, such as CDKN1C and PHLDA2, both expressed from the maternal chromosome. The centromeric ICR (IC2) corresponds to the promoter of the KCNQ1OT1 gene whose non-coding transcript silences the adjacent imprinted genes on the paternal chromosome. Loss of IC2 methylation on the maternal chromosome results in KCNQ1OT1 activation and CDKN1C silencing, and is the most frequent cause of BWS. Duplications of the entire centromeric domain on the maternal chromosome results in a SRS-like phenotype, while deletions in the maternal chromosome result in BWS, confirming the importance of the maternally expressed genes in growth control.4–9 In the mouse, a targeted deletion of the orthologous IC2 region causes activation of the maternally expressed genes and growth deficiency when paternally inherited.10 Unexpectedly, paternal 11p15.5 deletions have, up till now, been associated with normal phenotype in humans, indicating the complexity of imprinting control in this chromosomal region.6 ,8
Here, we describe a family in which two fetuses which died in utero with severe growth deficiency carried a paternally inherited 60 kb deletion including the IC2 region. The deletion resulted in loss of KCNQ1OT1 expression and CDKN1C and PHLAD2 activation. While this finding demonstrates a role of the centromeric 11p15.5 imprinted domain in intrauterine growth restriction (IUGR), comparison with previously described paternal 11p15.5 deletions provides important information on the location of the cis-acting regulatory elements of this chromosomal region.
Fetuses 1 and 2 were conceived by a phenotypically normal couple. They have previously given birth to a healthy child. In the following pregnancy, IUGR was noted: at 23 weeks, the fetus was compatible with 19 weeks gestational age. Doppler studies demonstrated absence of end-diastolic flow (AEDF) in the umbilical arteries which led to fetal demise at 27 weeks. The fetal weight was 365 g, and the placenta weighed 85 g. No malformations were noted. Laboratory tests ruled out any underlying maternal thrombophilia. In the subsequent pregnancy, the same clinical picture was manifested: fetus 2 showed delayed growth, and at 21 weeks was compatible with 19 weeks. Disturbed umbilical blood flow with AEDF and severe IUGR led to fetal demise at 27 weeks. The fetal weight was 360 g.
A microdeletion at chromosome 11p15.5 of 40–70 kb was first detected by chromosome microarray (CMA) (performed at Signature Genomics, Spokane, WA, using the Roche NimbleGen 135K oligonucleotide array as platform, and the SignatureChipOS Lot#: 528428A05 V.3.0 for the data analysis) in DNA extracted from amniocytes of fetus 2 (not shown). Fluorescence in situ hybridisation analysis of metaphase cells using the 11p15.5 BAC probe RP11-1061N1 (and the 11q12.1 probe RP11-872D17) showed one normal signal and one diminished signal in all cells examined (not shown), confirming the deletion. To assess the parental origin, and to determine more precisely the extent of the deleted region, the DNA of fetus 2 and the parents was further analysed by CMA using the Affymetrix Genome Wide Human Single Nucleotide Polymorphism (SNP) Array 6.0 and the Affymetrix Genotyping Console 3.0.2 software, as described.11 This analysis demonstrated the presence of a 52/58 kb deletion spanning from CN_582601 (Chr11: 2,684,979 on hg19) to CN_584828 (Chr11: 2,737,280) in the DNA of fetus 2 (figure 1A). Normal copy number was detected in parental DNAs with this technique. The genotypes derived from SNPs included in the deletion indicated that the mutation was on the paternally inherited chromosome (figure 1B). The amount of available DNA from fetus 1 was insufficient to perform a similar analysis. We, therefore, further refined the deletion boundaries on the DNA of fetus 2 by quantitative PCR, and used the obtained information to set up a PCR assay across the breakpoint. The primers were 5′-TGTTCAAGCTGTGGCCACTGG -3′ and 5′-GATGGAGTGTGGTGAGGCAC -3′ and the PCR conditions: 1.5 Mm MgCl2, 10% Dimethyl sulfoxide (DMSO), annealing temperature 60°C. By using this assay and DNA sequencing (from PRIMM, Italy), we demonstrated the presence of the microdeletion also in fetus 1 (figure 1C). The sequencing results demonstrated that the deletion spans from chr11:2679858 to chr11:2739436 (based on University of California Santa Cruz (UCSC) 2009 hg19 assembly), and includes the exon 11 of the KCNQ1 gene, the centromeric-imprinted control region (IC2) of the 11p15.5 imprinted gene cluster and the most 5′ 40 kb of the KCNQ1OT1 non-coding RNA gene (figure 1A). When parental DNAs were tested by PCR, a weak band corresponding to the region encompassing the deletion breakpoint was observed in the father, indicating somatic mosaicism (see online supplementary figure and supplementary data). All the cell and DNA samples investigated have been obtained after release of informed consent. Unfortunately, we could not analyse the DNA of the healthy sibling (II-1) because the parents did not consent for this test. However, we observe that the 11p15.5 60 kb deletion is not present in 1500 samples analysed with the same platform, as well as in the Database of Genomic Variants (http://projects.tcag.ca/variation/ and data not shown).
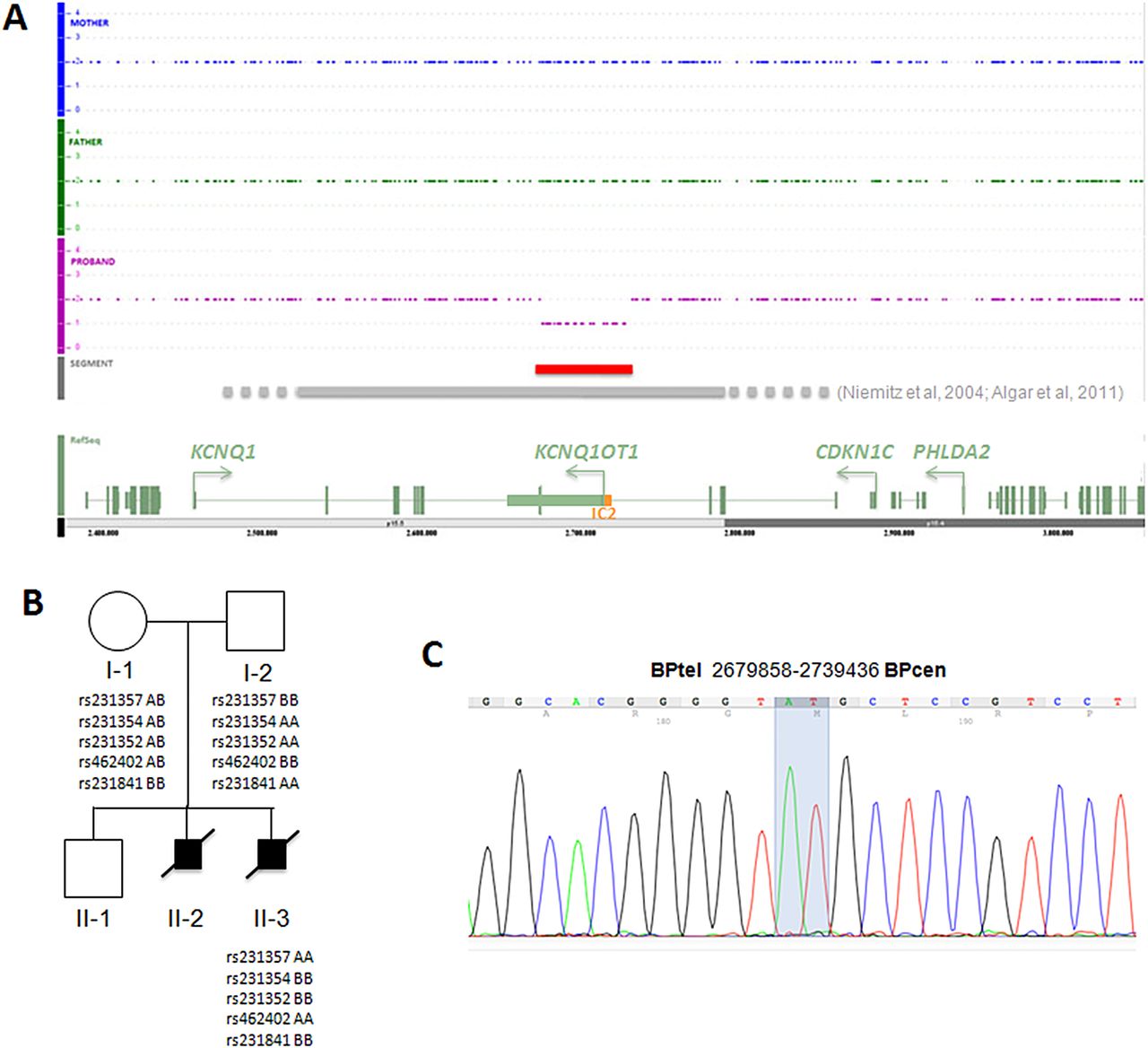
Structural characterisation of the 11p15.5 microdeletion. (A) Genomic profiles of fetus 2 and the parents at chromosome 11p15.5 as determined by SNP array. The extension of the deletion described in this study is indicated by a red line; those of previously described deletions are indicated by a grey line: the dotted ends represent the undefined borders of these deletions. A schematic diagram showing the relevant genes of the region is present at the bottom. The green rectangles represent the exons, the arrows indicate the transcription orientation, the orange box the IC2. (B) Pedigree of the family with relevant 11p15.5 genotypes. Fetus 1 and fetus 2 are indicated as II-2 and II-3, respectively. The informative SNPs present in the deleted region in the individuals investigated are shown. (C) Electropherogram showing the DNA sequence of the deletion breakpoint in fetus 1. The extreme nucleotides at the breakpoint are highlighted.
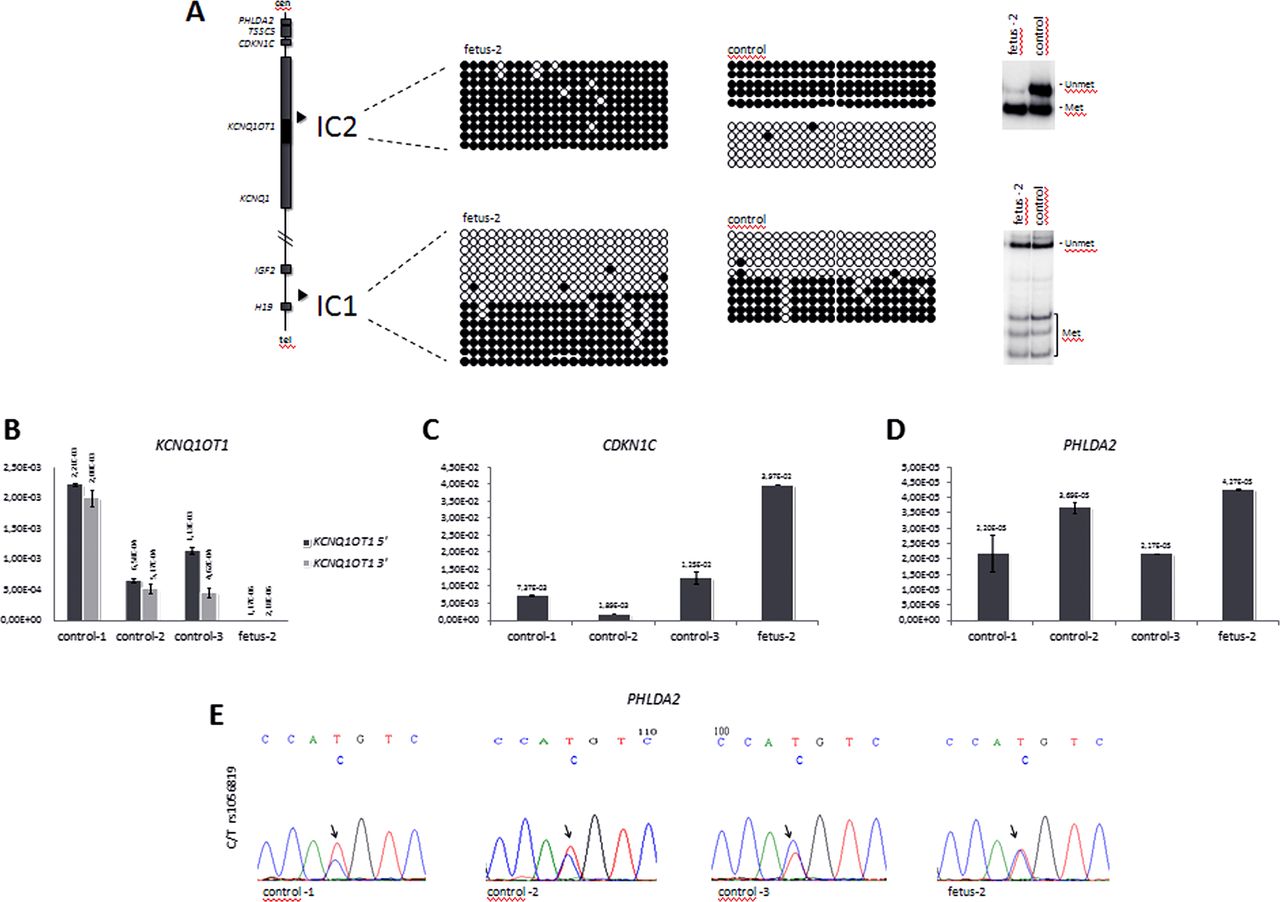
IC2 is normally methylated on the maternal chromosome and non-methylated on the paternal chromosome. We analysed DNA methylation by bisulphite sequencing and COmbined Bisulfite Restriction Analysis (COBRA), as described,11 and found that this region is differentially methylated on the maternal and paternal alleles in amniocytes from a normal control, but is completely methylated in the amniocytes of fetus 2 (figure 2A), thus confirming that the deletion is on the paternal chromosome and indicating the imprinting defect. A similar analysis performed on IC1 demonstrated DNA methylation levels similar to controls, thus showing that the 60 kb deletion does not affect the imprinted methylation of the telomeric 11p15.5 imprinted domain.
{kind=link}
{kind=link}
DNA methylation and gene expression analyses. (A) DNA methylation analysis of IC1 and IC2 in fetus 2 amniocytes by bisulphite sequencing and COBRA. A diagram representing the main genes and the positions of the ICs of the 11p15.5 cluster is shown on the left. For bisulphite sequencing (middle), DNA samples of fetus 2 and control amnyocites were treated with sodium bisulphite, amplified by PCR with IC1- or IC2-specific primers, cloned and sequenced; 23 CpGs of IC2 and 23 CpGs of IC1 (CCCTC-binding factor (CTCF) target site 6) are shown. Each line corresponds to a single template DNA molecule, and each circle represents a CpG dinuclotide. Filled circles designate methylated cytosine, and open circles correspond to unmethylated cytosines. For COBRA (right), 1 µg DNA was treated with sodium bisulphite, PCR-amplified and incubated with the restriction enzyme BstUI, as described.19 The slow-migrating bands correspond to the non-methylated allele, and the fast-migrating bands correspond to the methylated allele. Note, that IC2 is hypermethylated, consistent with the deletion of the non-methylated paternal allele, while IC1 methylation is normal. (B–D) Gene expression analysis. Levels of KCNQ1OT1 (B), CDKN1C (C), and PHLDA2 (D), RNAs were measured by quantitative real-time RT-PCR in fetus 2 and control amniocytes. Values were normalised against those of GAPD. Note, that KCNQ1OT1 is silenced, while CDKN1C and PHLDA2 are activated in the amniocytes of fetus 2. (E) Allele-specific expression of PHLDA2. 1 μg RNA derived from the amniocytes of fetus 2, and three controls was retrotranscribed. The PHLDA2 cDNA was PCR-amplified and sequenced. The electropherograms show the DNA sequence around SNP rs1056819. Note, that the two parental PHLDA2 alleles are equally expressed in fetus 2, but only the biased expression of one allele is present in the controls.
The IC2 region includes the promoter of the KCNQ1OT1 gene. The non-coding KCNQ1OT1 RNA is normally transcribed from the paternal chromosome, and represses the expression of CDKN1C and PHLDA2 in cis. We tested KCNQ1OT1, CDKN1C and PHLDA2 expression in fetus 2 and three control amniocyte lines by quantitative RT-PCR. cDNA was synthesised using 1 μg total RNA, and the Quantitech Reverse Transcription Kit (Qiagen) and Q-PCRs were run on a CFX96 Real-Time System+C1000 Thermal Cycler (Biorad) by using the iQ SYBR Green Supermix (BioRad). The PCR primers were: for the KCNQ1OT1 5′ end, 5′-CCTGATCCATGCAGCATGTTA-3′ and 5′-GTCCGTAAAATTGGAAGCCATT-3′; for the 3′ end, 5′-AGTCACATATAAGGGAATCAACAGC-3′ and 5′-ATTTCTGAAGGATAGCTTTGCTGGG-3′; for PHLDA2, 5′-CACGCCATGAGGCCATAC-3′ and 5′-GCACGGGAAGTTCTTCTGCT-3′. The primers for CDKN1C are already reported.11 The values were normalised against the expression of GAPD, as described.11 We found that the level of KCNQ1OT1 RNA in the amniocytes of fetus 2 was three orders of magnitude lower than that of controls (figure 2B). Conversely, CDKN1C expression was significantly increased (p<0.01) in fetus 2 when compared with the control amniocytes (figure 2C). PHLDA2 was weakly expressed; however, its mRNA level was found significantly higher (p<0.05) than that of two of the three control amniocyte lines investigated (figure 2D). Heterozygosity for SNP rs1056819 in the PHLDA2 gene was found in fetus 2 and allowed to determine its imprinted status. Sequencing of PHLDA2 cDNA (PCR primers: 5′-GTGACTACAAAGAACCAGCG-3′ and 5′-GCTCATCGATTTCCAGAACCG-3′) derived from amniocyte RNA demonstrated biased expression versus one of the parental alleles in the three normal controls, but equal expression of the maternal and paternal alleles in fetus 2, indicating loss of imprinting (figure 2C). The allele-specific expression of CDKN1C could not be tested because fetus 2 was not informative (not shown).
While maternal deletions of the centromeric domain of the 11p15.5 imprinted gene cluster are usually associated with BWS, paternal deletions have been demonstrated so far only in individuals with normal phenotype.9 In this report, we demonstrate that a paternally inherited 60 kb deletion encompassing the centromeric IC at 11p15.5 is associated with recurrent severe IUGR. We found that the deletion results in silencing of the non-coding KCNQ1OT1 gene, and activation of CDKN1C and PHLDA2. These results demonstrate that the phenotype associated with 11p15.5 deletions is strongly influenced by the extent of the region involved, and indicate defective imprinting of the centromeric domain as a mechanism underlying severe prenatal growth restriction. Since the deletion was detected in the father by PCR but not CMA analysis, and did recur in two of his fetuses, we hypothesise that the father has somatic as well as germ-line mosaicism for the deletion. Due to the significant recurrence risk, the parents are now considering preimplantation genetic diagnosis.
In the mouse, targeted deletion of the orthologous IC2 region results in growth restriction and loss of imprinting of six genes including Cdkn1c and Phlda2 when paternally inherited.10 By contrast, paternal transmission of 250–330 kb deletions, including most of the KCNQ1 gene, IC2 and KCNQ1OT1 is associated with normal phenotype in humans (refs. 6 and 8; also see figure 1). Because the existence of distal enhancers for Cdkn1c were demonstrated in the mouse,12 ,13 it was proposed that these cis-acting elements were removed together with KCNQ1OT1 by the 250–330 kb deletions, thus preventing activation of the paternal CDKN1C allele.6 ,8 For the same reason, maternal transmission of these deletions likely results in CDKN1C silencing and BWS. PHLDA2 is imprinted with incomplete repression of the paternal allele in fetal tissues.14 We demonstrated silencing of KCNQ1OT1 and equal expression of the maternal and paternal PHLDA2 alleles in the amniocytes of fetus 2, suggesting that paternal transmission of the 60 kb deletion releases the imprinted repression of the centromeric domain genes by abolishing KCNQ1OT1 promoter and expression. The increased CDKN1C and PHLDA2 mRNA levels detected in fetus 2 are also consistent with loss of imprinting, and indicates that the enhancers for CDKN1C and PHLDA2 are not included in the 60 kb deletion, thus explaining the growth restriction phenotype.
CDKN1C (also known as p57KIP2) is an inhibitor of G1 cyclin-dependent kinases, and a negative regulator of cell proliferation.15 A role of CDKN1C in IUGR was recently demonstrated by the finding of gain of function mutations in the IMAGe syndrome.16 Maternal duplications likely increasing CDKN1C expression were found in individuals affected by SRS.4 ,5 PHLDA2 is a tumour suppressor gene and regulates placental and in utero fetal growth.17 ,18 Although hypomethylation of the ICR of the 11p15.5 telomeric domain (IC1) is a common cause of SRS, imprinting defects of the centromeric domain have never been described.3 The phenotype associated with the 60 kb deletion suggests that loss of imprinting of CDKN1C, PHLDA2 and, possibly, other genes of the centromeric domain, leads to such a severe growth deficiency as to cause fetal demise. It may therefore be important to look for such epigenetic defects in the cases of fetal death with severe IUGR.
Acknowledgments
We thank the family for their cooperation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figure
Footnotes
-
Contributors FC, AR, YY: study design, data analysis and interpretation and manuscript writing and revision. ADC, AS, OP, MC, MM: performed cell culture and molecular analyses. MB, YY: patient recruitment, examination and counselling.
-
Funding This work was supported by grants from MIUR PRIN 2009 (to AR), Telethon-Italia grant no. GGP11122 and Progetto Bandiera MIUR-CNR Epigenomica (to AR), and Associazione Italiana Ricerca sul Cancro (to FC and AR).
-
Competing interests None.
-
Patient consent Parental consent obtained.
-
Ethics approval Comitato Etico della Seconda Università di Napoli-Azienda Policlinico.
-
Provenance and peer review Not commissioned; externally peer reviewed.