Article Text

Abstract
Background Deregulation of the activity of the ubiquitin ligase E6AP (UBE3A) is well recognised to contribute to the development of Angelman syndrome (AS). The ubiquitin ligase HERC2, encoded by the HERC2 gene is thought to be a key regulator of E6AP.
Methods and results Using a combination of autozygosity mapping and linkage analysis, we studied an autosomal-recessive neurodevelopmental disorder with some phenotypic similarities to AS, found among the Old Order Amish. Our molecular investigation identified a mutation in HERC2 associated with the disease phenotype. We establish that the encoded mutant HERC2 protein has a reduced half-life compared with its wild-type counterpart, which is associated with a significant reduction in HERC2 levels in affected individuals.
Conclusions Our data implicate a model in which disruption of HERC2 function relates to a reduction in E6AP activity resulting in neurodevelopmental delay, suggesting a previously unrecognised role of HERC2 in the pathogenesis of AS.
- Developmental
- Clinical genetics
- Neurosciences
Statistics from Altmetric.com
Introduction
Angelman syndrome (AS, (MIM 105830)) and Prader–Willi syndrome (PWS, (MIM 176270)) are distinct neurodevelopmental conditions caused by abnormalities at the 15q11–q13 imprinted region. Both conditions present with characteristic developmental, neurological and behavioural phenotypes. Most commonly these disorders result from a 5–7 Mb deletion of the imprinted region, which is either paternal in origin (PWS) or maternal in origin (AS), with clustered breakpoints at either of two centromeric sites and one telomeric site.
Homologous recombination between chromosome-specific low-copy repeats (duplicons) is the mechanism underlying a number of genetic disorders.1 Sequences homologous to the HERC2 (HECT domain and RCC1-like domain 2, MIM 605837, NM 004667.4) gene, encoding a giant 528 kDa protein, form the basis of the low-copy repeats associated with the 15q11–15q13 breakpoints. Evolutionary evidence is suggestive of a HERC2 duplicon-mediated process leading to an increased tendency for chromosomal rearrangements to occur in this region. Although AS and PWS deletion patients are hemizygous for HERC2, to date there has been no evidence that this gene contributes to either phenotype.2–5
Patients with classical AS present with severe mental retardation and profound speech impairment with only minimal use of words, although non-verbal and receptive communication skills are higher than expressive language skills. A movement disorder ranging from ataxia to clumsiness or unsteadiness and a characteristic behaviour pattern of hyperactivity, a happy personality and episodes of inappropriate bursts of laughter are typically seen. A wide based gait with pronation at the ankles and arms held at shoulder level and flexed at the elbows are frequent findings. Hypotonia and feeding difficulties are common in infancy with increased tone in the lower limbs and hyperactive reflexes developing later. Seizures are present in over 80% of AS patients and comprise a diversity of types; an EEG pattern of 2–3 Hz large amplitude slow wave bursts is typical and seizures decrease in frequency in adulthood. Brain structure is normal by CT or MRI imaging.
Scoliosis, strabismus and hyperactivity are associated features of the condition. Dysmorphic features are subtle and include microcephaly (by the age of 2 years), prominent chin, macrostomia, hypopigmentation of skin, hair and eyes (deletion patients only). The clinical features of AS have been reviewed previously.6–9
Recently we demonstrated physical and functional interactions of HERC2 with E6AP, the primary molecule implicated in the pathogenesis of AS. Most significantly HERC2 was shown to stimulate the E3 ubiquitin-protein ligase activity of E6AP implicating it as a regulator of E6AP and raising the intriguing possibility that HERC2 may play an important role in the AS phenotype.10 In the current study, we investigated a unique and recognisable inherited neurodevelopmental condition with some phenotypic similarities to AS, present at high frequency among the Ohio Old Order Amish community, which we show to be due to HERC2 mutation.
Methods
Subjects
Blood or buccal samples were obtained from affected children, parents and unaffected siblings. DNA and RNA were extracted by standard procedures. A single 0.4 mm diameter skin biopsy was taken from affected individuals XI:2, XI:3, X:I and X:2 and from unaffected individuals. All samples were obtained with approved informed consent (University of Arizona IRB00000291).
Genotyping and linkage analysis
Single-nucleotide polymorphism (SNP) microarray genotyping was performed using Illumina Human CytoSNP-12v2.0 and 2.1 330 K arrays. Marker saturation was carried out using existing and newly generated microsatellite markers (primer sequences available on request). Alleles were size-fractionated using 8% polyacrylamide gels and DNA was visualised by silver staining. Multipoint linkage analysis performed with Simwalk211 under a model of autosomal recessive inheritance with full penetrance, using a disease allele frequency estimated at 0.0003. The pedigree was deconstructed to facilitate computational efficiency.
Mutation analysis
Unique intronic primers were designed for 52 HERC2 exons as well as exons of other genes located in the critical interval. The repetitive nature of HERC2 prohibited amplification from genomic DNA; as such the remaining 40 exons were amplified from cDNA using unique exonic primers. Primers were designed using online Primer3 software based on sequences from the online University of California, Santa Cruz (UCSC) Genome Browser database and ordered from Sigma-Aldrich (optimum PCR conditions are available on request). RT-PCR was carried out using PrimeScript One Step RT-PCR Kit V.2, TaKaRa according to the manufacturer's instructions.
Purified PCR amplification products were sequenced using dye-terminator chemistry and electrophoresed on an ABI3130 XLA capillary sequencer (Applied Biosystems). Mutation analysis was carried out using Finch TV 1.4.0 (created by Geospiza Inc.) and Gene Tool 1.0.0.1 (created by Bio Tools Inc). Both strands of each product were sequenced.
Seventy nine anonymised regional control and 169 European control DNA samples were screened by bidirectional sequencing for HERC2 exon 13.
Cell lines, plasmids and transfections
Primary fibroblasts, HEK293T cells and U2OS cells were cultured in DMEM (Sigma Aldrich) with 10% fetal bovine serum (FCS), 2 mM Glutamine (Invitrogen) and 1% penicillin and streptomycin.
Expression constructs (transient transfection experiments) encoding HA-tagged wild-type (wt) E6AP (isoform 1), the HA-tagged catalytically inactive p.Cys820Ala mutant of E6AP, Myc-tagged p.Ile53Ser Ring1B mutant, HA-tagged HERC2 and His-tagged ubiquitin were described previously;12 Myc-HERC2 (1–1295) was kindly provided by T. Ohta. The cDNAs encoding the p.Pro594Leu HERC2 mutants (full-length or amino acid residues: 1–1295), were generated by a PCR-based approach (further details will be provided upon request) and expressed as N-terminally HA-tagged or Myc-tagged forms from pcDNA3. For transient expression, HEK293T and U2OS cells were transfected with the respective constructs in the presence of a reporter construct encoding β-galactosidase by lipofection (Lipofectamine 2000 or LTX) according to the manufacturer's instructions (Invitrogen). Protein extracts were prepared 24–48 h after transfection as described13 and transfection efficiency was determined by measuring β-galactosidase activity. Extracts were analysed by Polyacrylamide gel electrophoresis (PAGE) and immunoblotting as previously described.13 Protein levels were normalised and expressed as percentage of controls. The antibodies used were: anti-HERC2 (BD Biosciences); anti-E6AP (Santa Cruz Biotechnology, Inc); anti-XPA (Kamiya Biomedical Company); anti-Myc (Roche); anti- α tubulin (Calbiochem); anti-Ran; horseradish peroxidase-conjugated secondary antibodies (Invitrogen).
Ubiquitination and degradation assays
For ubiquitination of p.Ile53Ser Ring1B mutant within cells, one 6 cm plate of HEK293T cells was transfected with expression constructs encoding HA-tagged p.Ile53Ser Ring1B mutant (1 µg), E6AP (1.6 µg), His-tagged ubiquitin (1.5 µg) and HERC2 or the p.Pro594Leu HERC2 mutant (3.5 µg, 2.5 µg or 1.5 µg) as indicated (figure 4). Twenty-four hours after transfection, 30% of the cells were lysed under non-denaturing conditions to determine expression levels of E6AP, p.Ile53Ser Ring1B mutant and the two forms of HERC2. The remaining cells were lysed under denaturing conditions, and ubiquitinated proteins were purified. For cycloheximide assays using ectopically expressed HERC2 and p.Pro594Leu HERC2 mutant, 107 HEK293T cells were transfected with 10 µg of the HERC2 expression constructs indicated. Twenty-four hours post transfection, cells were trypsinised, pooled and equal aliquots seeded onto 8×6 cm plates. After additional 24 h, cells were treated with 60 µg/ml cycloheximide (dissolved in MeOH) in the absence or presence of 10 µM MG132 (dissolved in Dimethyl sulfoxide (DMSO)) and harvested at the times indicated. Protein extracts were prepared and analysed by SDS-PAGE followed by western blot analysis. Levels of the various HA-tagged proteins (E6AP, p.Cys820Ala E6AP mutant, HERC2 and p.Pro594Leu HERC2 mutant) and Myc-tagged p.Ile53Ser Ring1B mutant were determined by western blot analysis. The antibodies used for detection were the mouse monoclonal HA11 (Hiss Diagnostics, Freiburg, Germany) for HA-tagged proteins and the mouse monoclonal 9E10 (Abcam) for Myc-taggedp.Ile53Ser Ring1B mutant protein.
Results
Fifteen affected individuals, aged between 11 months and 39 years, were identified (figure 1). Cardinal features include global developmental delay, hypotonia, delay in ambulation between ages 3–5 years, an unstable gait frequently with a broad base and arms held upwards and bent at the elbows with brisk walking or running. Failure to acquire normal speech was present in all affected individuals. Those with extreme poverty of speech use gesturing and communication aids effectively and understanding is far greater than language ability. Seizures were reported in 4/15 affected cases. Neuroimaging was only available for 6/15 individuals, in three cases absence of the posterior half of the corpus callosum was reported, the remaining three were said to be unremarkable.

(A) and (B) Pedigree drawings of the two Amish families.
Affected individuals were frequently reported to have poor concentration and hyperactivity. Dysmorphic features are subtle and include plagiocephaly, prognathism, narrow palate, elongated hallux, sandal gap and excessive pronation of the feet occasionally requiring surgery. The clinical features are detailed in tables 1A and 1B.
A comparison of the clinical findings of individuals homozygous for HERC2 c.1781C>T (Pedigree 1A)
A comparison of the clinical findings of individuals homozygous for HERC2 c.1781C>T (Pedigree 1B)
Interestingly the majority of affected individuals in the Amish community have bright blue eyes due to the coincident close proximity of the causative founder mutation to a SNP rs12913832 in HERC2 that influences OCA2 expression and is associated with eye colour (data not shown).14
Assuming that a founder mutation was responsible we undertook a genome wide microarray scan using Illumina Human CytoSNP-12v2.0 and 2.1 330K arrays using DNA from the affected individuals initially available (VIII:7, VIII:8, IX:6, IX:7, X:5, X:6). This identified a single notable homozygous region shared by all affected individuals of 1.8Mb on chromosome 15q13.1–q13.3, delimited by markers rs1378094 and rs10162860, likely to correspond to the disease locus. Subsequent microsatellite marker analysis (see online supplementary figure S1) confirmed autozygosity across this region and refined and precisely positioned the disease locus (LODmax 12.5). The putative disease locus is predicted to contain 28 transcript-encoding regions, of which three are hypothetical, two are microRNAs and 17 are pseudogenes. While screening the remaining six genes a non-synonymous base variant was identified in exon 13 of HERC2 (NM_004667.5:c.1781C>T), predicted to encode a HERC2 protein with substitution of proline by leucine at amino acid position 594 (see online supplementary figure S2). This substitution is not a known SNP and is not present in the 1000 Genomes Project or the Exome Variant Server, NHLBI GO Exome Sequencing Project (6503 subjects). Genotyping of the full family revealed that the sequence variant cosegregated appropriately for a recessive disorder among the entire extended family comprising seven nuclear families, with affected individuals being homozygous, parents being heterozygous carriers and unaffected siblings being either wt or heterozygous carriers of the variant (figure 1A). The c.1781C>T variant was also found to cosegregate in a second Amish family comprising four affected individuals from a distinct Amish deme (figure 1B). Analysis of 158 control chromosomes from unaffected individuals from the same Ohio Amish community identified two heterozygous carriers of the sequence variant. RNA was extracted from the whole blood of two of the affected cases (IX:6 and IX:7) for cDNA synthesis and sequencing, which verified the presence of the variant in the RNA transcript.
Given the proximity of the HERC2 gene to the AS/PWS imprinting control region, we analysed methylation status at the SNRPN imprinted locus by methylation specific PCR; no abnormalities were detected (data not shown).
We next investigated the functional consequences of the HERC2 mutation. Western blot analysis revealed a profound effect of the mutation on protein levels, with HERC2 protein being almost undetectable in fibroblasts derived from four affected individuals in comparison with three healthy controls. This effect seems specific for HERC2 because protein levels of known HERC2 interacting proteins such as E6AP or XPA, or the unrelated protein α-tubulin were not significantly altered (figure 2A). The dramatic reduction of HERC2 protein levels may relate to reduced expression at the mRNA level associated with reduced transcription rate or mRNA stability, or reduced stability of the translated protein product. We therefore evaluated HERC2 mRNA levels by quantitative PCR and tested the effect of a proteasome inhibitor cocktail on p.Pro594Leu HERC2 levels in patient primary fibroblasts. This revealed no significant alteration in mRNA levels (data not shown). Evaluation of HERC2 levels in patient fibroblasts following treatment with proteasome inhibitors was suggestive of increased turnover, although this could not be established conclusively due to limited availability of suitable patient cellular material (data not shown). Consequently, we analysed the turnover of the mutant protein by transiently expressing wt HERC2 as well as p.Pro594Leu mutant HERC2 in HEK293T cells. This revealed more rapid degradation of the p.Pro594Leu HERC2 mutant (figure 2B) indicating that the mutant protein has a shorter half-life than wt HERC2.
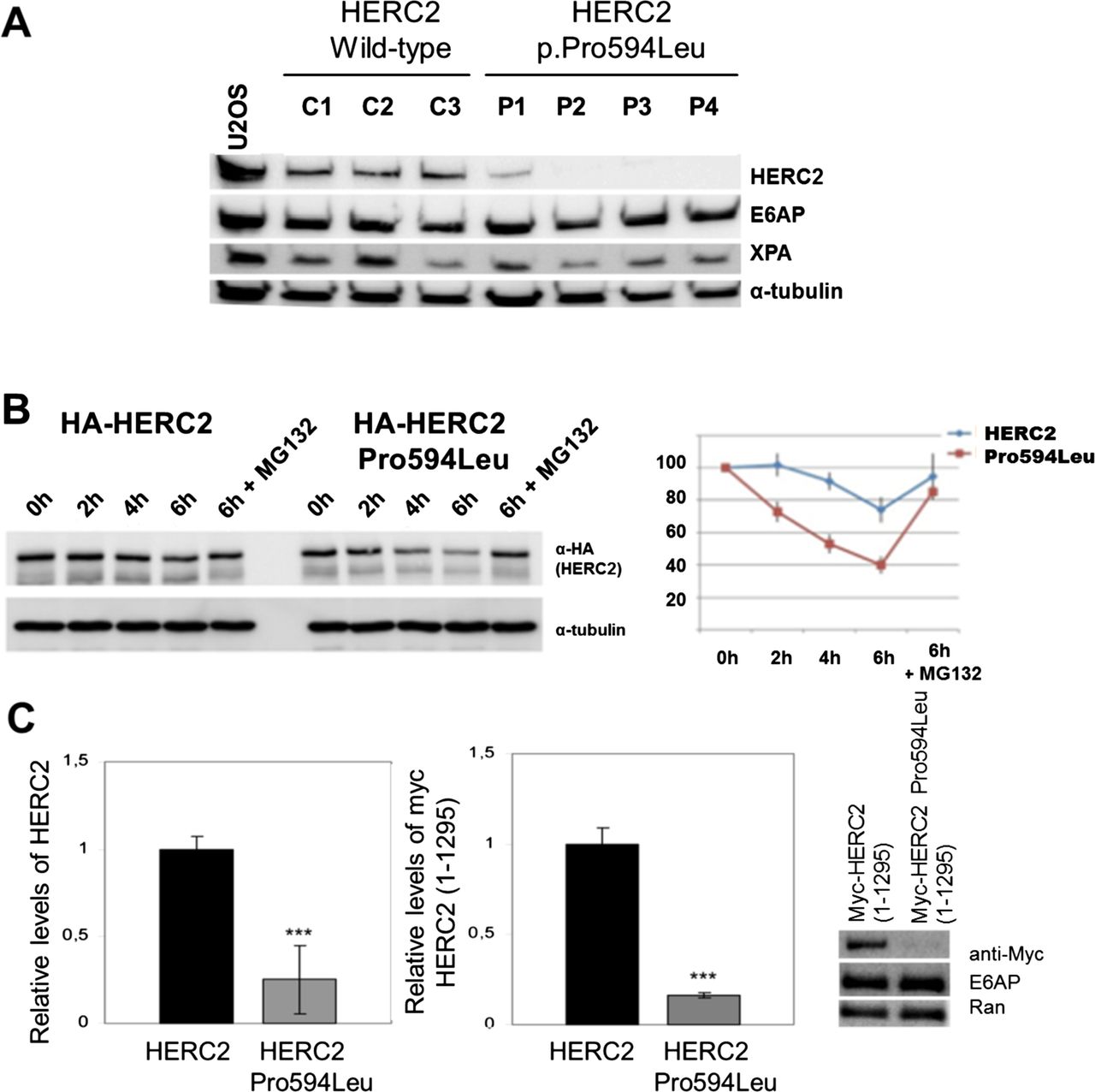
(A) Levels of HERC2 are diminished in fibroblasts from affected individuals. Lysates from fibroblasts of wild-type (wt) controls and affected individuals were analysed by immunoblot with the indicated antibodies. The levels of HERC2 were normalised with respect to α-tubulin as is indicated in the right panel. (B) The p.Pro594Leu HERC2 mutant has a shorter half-life than the wt protein. HEK293T cells with a stable knockdown of endogenous HERC2 expression (unpublished) were transfected with expression constructs encoding HA-tagged forms of wt HERC2 and p.Pro594Leu mutant HERC2. After 48 h, cells were treated with cycloheximide in the absence or presence of 10 µM MG132, harvested at the times indicated, and protein levels analysed by western blot analysis using the antibodies indicated. (C) U2OS cells were transfected with the indicated constructs and analysed by immunoblot. The right panel shows the levels of expression with respect to Ran.
A possible explanation for the apparent instability of the p.Pro594Leu HERC2 mutant may be provided using structural data from the protein data bank.15 HERC2 is a member of the RCC1 (Regulator of Chromosome Condensation 1) superfamily, each of which contains at least one RCC1-like domain (RLD). Within its 4834 amino acid sequence, HERC2 contains three RLD domains and a C-terminal HECT domain (homologous to E6AP carboxy terminus) with E3 ligase activity. The p.Pro594Leu amino acid substitution affects a proline residue of the RLD1 domain of HERC2 highly conserved in all species examined (see online supplementary figure S3A,B) and among all human RCC1 superfamily proteins.16 ,17 All RLDs are thought to consist of a seven-bladed β-propeller structure similar to RCC1.18 Although no structural data is available for RLD1 of HERC2, given its similarity to RLD3,17 it is highly likely to form such a β-propeller structure. In silico superposition of RCC1 and predicted HERC2 RLD3 structures (Jmol/SuperPose/FirstGlance) permitted us to extrapolate the position of P594 in the RLD1 domain of HERC2 to the fourth propeller blade (figure 3). Substitution of the highly conserved Pro594, located within the fourth blade of the propeller, is likely to disrupt the loop between the fourth and the fifth blades and may result in a conformational change affecting protein structural stability and lead to increased turnover rate. To investigate this further, we transfected U2OS cells with wt Myc-HERC2 (residues 1–1249) as well as p.Pro594Leu mutant Myc-HERC2 (residues 1–1249) gene constructs. This confirmed a profound reduction of protein levels associated with substitution of Pro594 (figure 2C), further evidence that protein instability is the likely cause of reduced HERC2 levels in patient fibroblasts. In light of the observation that HERC2 stimulates E6AP, we next examined the effect of the p.Pro594Leu HERC2 substitution on E6AP E3 ligase activity. Given that known substrates of E6AP are either not detectable in primary fibroblasts (activity-regulated cytoskeleton-associated protein (ARC); unpublished data) or particularly short-lived (Ring1B),19 we studied E6AP-mediated ubiquitination of a Ring1B mutant in transient transfection experiments. This revealed that wt HERC2 as well as p.Pro594Leu HERC2 mutant stimulate E6AP-mediated ubiquitination of Ring1B in a dose-dependent manner (figure 4), however mutant HERC2 was found to be less active than wt HERC2. Similar results were obtained for autoubiquitination of E6AP (data not shown). Although the E6AP activation difference detected was moderate, and may not in itself be functionally significant, the dose-dependent nature of HERC2 on E6AP activation is likely to exacerbate effects of the p.Pro594Leu HERC2 mutation.

Superposition of Human RCC1 (cyan) and human HERC2 RLD3 (light blue) Lateral (A,C,D) and bottom (B) views of the β-propeller. High conserved proline, equivalent to proline 594 of HERC2, is shown in RCC1 (yellow) and in RLD3 (red). The N-termini and C-termini of RCC1 are indicated. The figure was generated and edited using the SuperPose and FirstGlance in Jmol programmes. Protein Data Bank entries: 1A12 for RCC1 and 3KCI for HERC2 RLD3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of p.Pro594Leu HERC2 on E6AP-mediated Ring1B ubiquitination. HEK293T cells with a stable knockdown of endogenous HERC2 expression (unpublished) were co-transfected with expression constructs for His-tagged ubiquitin, Myc-tagged p.Ile53Ser Ring1B mutant, HA-tagged E6AP, and HA-tagged full-length versions of HERC2 or p.Pro594Leu mutant HERC2 as indicated. 24 h after transfection, protein extracts were prepared and ubiquitinated proteins were isolated by Ni2+-affinity chromatography. Upon purification, levels of ubiquitinated p.Ile53Ser Ring1B mutant protein were determined by western blot analysis with an anti-Myc antibody (upper panel). Input, corresponds to 20% of the proteins extracts used for affinity purification. *, monoubiquitinated p.Ile53Ser Ring1B mutant protein.
Discussion
Taken together our genetic and cellular data implicate HERC2 in a human disease phenotype which, although a distinct entity, has some AS-like features.
There are 18 reported members of the human RCC1 protein superfamily, six of these within the HERC subgroup. A number of proteins within this family have been associated with a diverse set of human diseases,17 ,20 until now however no HERC subgroup members had been causally linked. Human and mouse HERC2 protein display 95% homology and 99% similarity5 and HERC2 homozygous and hemizygous mutations have been identified as the cause of the mouse rjs syndrome (runty, jerky, sterile) also known as jdf2 (juvenile development and fertility-2). Affected mice are 20% smaller than their normal counterparts from birth, have abnormal gait and display behavioural abnormalities,21 reflecting similarities with features seen in our affected subjects. None of the affected individuals that we have seen have shown any significant differences from their healthy siblings in terms of birth weight or subsequent growth, nor have any pubertal abnormalities been reported. We have not observed any reduction in life expectancy; however the oldest affected individual we have seen is 39 years old. There does however appear to be some cognitive and behavioural decline in two of our oldest patients. Currently there is no mouse model of p.Pro594Leu HERC2, and production of such a future model may allow a more direct phenotypic comparison.
All AS cases associated with UBE3A mutations reported to date are predicted to affect the E3 ligase activity of E6AP.7 The previously reported interaction between HERC2 and E6AP results in stimulation of E6AP E3 ligase activity and involves HERC2 RLD2,10 whereas the variant reported here affects RLD1. Our data indicate a profound effect of the p.Pro594Leu mutation on HERC2 protein levels, which likely relate to reduced half-life resulting from distortion of the structure of the HERC2 molecule. The modest effect on E6AP stimulation associated with the HERC2 p.Pro594Leu mutation described here may in part explain why the developmental difficulties observed in our subjects are not as profound as those seen in AS patients in whom there is likely a more significant loss of E6AP activity. Seizures are reported in excess of 80% of AS patients. Although seizure activity was only reported in 4/15 subjects, we speculate that this may indicate that a subtle decrease in E6AP activity lowers the threshold for seizure activity. Microcephaly is not a feature of this condition, however this feature of AS is most commonly associated with microdeletions and less so with other molecular mechanisms leading to AS.9 Notable similarities between AS and the individuals with HERC2 mutation described here include hypotonia in childhood, a similar arms raised posture when walking particularly at speed, excessive pronation of the ankles, clumsiness and poor concentration. AS individuals, most commonly women, frequently develop scoliosis with age. Although we only observed three adult females, it is noteworthy that one had developed severe scoliosis. Strabismus is another feature commonly seen in AS and was reported in 4/15 affected individuals. Importantly a number of significant differences between the two conditions were also apparent. Although the parents of our Amish subjects did not report inappropriate bouts of laughter, many did remark on the sociability and loving nature of their children, however behavioural difficulties, aggression and stereotypies were also reported. Further distinguishing features include lack of the classical AS facial gestalt and no reports of ataxia. It is interesting that in three out of six of our subjects with neuroimaging data there was absence of the posterior part of the corpus callosum, which in association with the other clinical features described may be helpful in terms of reaching a diagnosis in patients with this condition. Finally although both conditions are characterised by significant speech abnormalities, AS individuals rarely develop any meaningful speech, whereas a number of individuals homozygous for the HERC2 mutation have developed the ability to communicate effectively with language. Good non-verbal communication skills and relatively preserved receptive language ability are features of both conditions. It was notable that those subjects who had received early and intensive speech and language therapy appeared to have made significantly more progress in language attainment.
In the current report, we describe a novel neurodevelopmental syndrome with some features reminiscent of AS arising due to HERC2 mutation. Although AS had not been suspected in any of the patients described here and we recognise that there are important differences between the two conditions, it remains an intriguing possibility that more deleterious mutations in the HERC2 gene may be detected in more severely affected patients who display a more significant phenotypic overlap with AS.
Acknowledgments
We are very grateful to the Amish families for partaking in this study, and to the Amish community for their continued support of the Windows of Hope project. We thank G. Pons for his help with protein structure programmes and T. Ohta for the Myc-HERC2 plasmid.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
GVH, ELB, HC contributed equally. SK, MC-R contributed equally. AHC, JLR, MS and BAC all contributed equally
-
Contributors AHC designed the genetics experiments. GVH, ELB, BAC and DJGM performed the genetics experiments. AHC, JLR, MS and ELB designed the functional cell biology experiments. SK, MCR, KM, JLR and MS performed the cell biology experiments. ELB, HC, DLF, MAP, KW and RC contributed clinical data to the manuscript. AHC, ELB, BAC, JLR and MS wrote the manuscript.
-
Funding The work was supported by MRC grant (G1002279), Newlife Foundation (AHC, GVH and ELB), MRC Clinical Research Training fellowship (G1001931 to ELB), the Deutsche Forschungsgemeinschaft (SCHE 346/6-1 to MS), and by MICINN (Ministerio de Ciencia e Innovación) grant (BFU2011-22498) to JLR. MCR is supported by a doctoral fellowship from Generalitat de Catalunya.
-
Competing interests None.
-
Ethics approval University of Arizona.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Any unpublished data from this study is available on request from the corresponding author.
-
Web references The URLs for data presented herein are as follows: Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/. ClustalW2, http://www.ebi.ac.uk/Tools/msa/clustalw2/. Jalview, http://www.jalview.org. Jmol, http://www.jmol.sourceforge.net. FirstGlance in Jmol, http://www.bioinformatics.org/firstglance/fgij/. SuperPose, http://www.wishart.biology.ualberta.ca/SuperPose/. Primer3, http://www.frodo.wi.mit.edu/. 1000 Genomes Project, http://www.1000genomes.org.Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA,: http://evs.gs.washington.edu/EVS/) (accessed, November, 2012).
-
Accession numbers The databank accession number for HERC2 reported in this paper is NM_004667.5.