Article Text

Abstract
The 11p15 region is organised into two independent imprinted domains controlled by imprinting control regions, which carry opposite germline imprints. Dysregulation of 11p15 genomic imprinting results in two human fetal growth disorders (Silver-Russell syndrome (SRS, MIM 180860) and Beckwith-Wiedemann syndrome (BWS, MIM 130650)) with opposite growth phenotypes. The mouse orthologous region on distal chromosome 7 (dist7) is well conserved in its organisation and its regulation. Targeted mutagenesis in mice has provided highly valuable clues in terms of the mechanisms involved in the regulation of genomic imprinting of the 11p15/dist7 imprinted region. On the other hand, the recent identification of unexpected genetic defects in BWS and SRS patients also brought new insights into the mechanisms of 11p15 imprinting regulation. However, some mouse models and human genetic defects show contradictions in term of growth phenotypes and parental transmission. In this review, we extensively analyse those various mouse and human models and more particularly models with mutations affecting the two imprinting centres, in order to improve our understanding of regulation of 11p15/dist7 genomic imprinting.
- Genomic imprinting
- Beckwith-Wiedemann syndrome
- Silver-Russell syndrome
- Fetal growth
- CTCF
Statistics from Altmetric.com
The 11p15/dist7 imprinted region
Structural organisation of the region
In both mice and humans, the region is organised into two imprinted domains, each of them under the control of its own imprinting control region (ICR): ICR1 (also called H19 DMR (differentially methylated region) or H19 DMD (differentially methylated domain) in the mouse) for the IGF2/H19 domain, and ICR2 (also called KvDMR1) for the CDKN1C/KCNQ1 domain (figure 1). Those domains include genes, such as the IGF2 and the CDKN1C genes, which are crucial for regulation of fetal growth. In several ways, the 11p15 region is an exemplary model of regulation of genomic imprinting. Firstly, the two ICRs carry opposite germline imprints: ICR1 is one of the few ICRs which are marked by DNA methylation in the male germline, whereas ICR2 is marked by DNA methylation in the female germline. Secondly, the two ICRs are differently regulated by two major mechanisms of imprinting control. ICR1 functions as an insulator whereas ICR2 serves as a promoter for a regulatory non-coding RNA (ncRNA), although recent data in mouse and human models suggest that other regulatory mechanisms are also at work for the CDKN1C/KCNQ1 domain.1–3

The human 11p15 region is organised in two imprinted domains, the IGF2/H19 and the CDKN1C/KCNQ1 domains, each of them under the control of its own imprinting centre, imprinting control region (ICR)1 and ICR2, respectively. ICR1 and ICR2 are methylated on the paternal and maternal allele, respectively (black circles). Some genes on both sides of the CDKN1C/KCNQ1 domain (ASCL2, CD81, TSSC4, OSBPL5) are imprinted only in placenta. The orthologous mouse region is very well conserved, except for the Insulin gene that is imprinted in mice but not in humans. This figure is only reproduced in colour in the online version.
ICR1 functions as a chromatin insulator and controls the reciprocal imprinting of the paternally expressed IGF2 and maternally expressed H19 genes. IGF2 and H19 are widely expressed during embryonic development and postnatally downregulated in most tissues. IGF2 encodes a protein that plays a major role in promoting embryonic and placental growth, whereas the exact function of the H19 ncRNA remained obscure until very recently (see below). ICR1 carries DNA methylation and repressive histone marks on the paternal allele and permissive histone marks on the maternal allele. The reciprocal imprinting of IGF2 and H19 occurs through the differential allelic binding (on the unmethylated maternal allele) of the zinc finger CTCF protein at four and seven binding sites on the mouse and the human ICR1, respectively. CTCF plays an important role in organising allele-specific higher order chromatin conformation, and cohesin has been showed to contribute to chromatin looping.4 Other nc RNAs such as miR-6755 or the long 91H antisense RNA6 have also been described within the IGF2/H19 domain. A very recent paper showed that the main function of H19 is in limiting growth of the placenta before birth, by releasing miR-675 which inhibits cell proliferation.7 Very recently, a new 6 kb gene called H19 opposite tumour suppressor (HOTS) was also identified in the human IGF2/H19 ICR1 domain.8 HOTS is expressed from the maternal allele, antisense to H19 and overlaps H19. It encodes a 150 amino acid nuclear protein with tumour suppressor function domain.8
ICR2 is an imprinting centre regulating imprinting of genes on either side. The imprinted CDKN1C/KCNQ1 domain covers approximately 400 kb in the embryo and 750–800 kb in the placenta (from ASCL2 to OSBPL5) (figure 1). ICR2 carries DNA methylation and repressive histone marks on the maternal allele and permissive histone marks on the paternal allele. ICR2 is located within the 10th intron of the long KCNQ1 gene and encompasses the promoter for a macro ncRNA (antisense KCNQ1OT1 RNA). The KCNQ1OT1 transcript is 80–120 kb long,9 although a recent study suggests it extends 470 kb from the transcription site in both embryonic and extra-embryonic tissues.10 It has a nuclear localisation and is first expressed in two-cell embryos. The KCNQ1OT1 RNA is expressed from the paternal allele and results in the paternal silencing of all imprinted cis-linked genes within the domain. The major mechanism of control of imprinting at ICR2 is achieved through the transcription of the full ncRNA by a mechanism which involves the recruitment of histone methyltransferases and repressive chromatin modifications (reviewed in Mohammad et al11). CTCF binding sites have also been identified at ICR2 on the unmethylated paternal allele,12 but how these CTCF binding sites regulate imprinting and chromatin looping is unknown.
Epigenetic marking of the region
Genomic imprints need to be reset at each generation. Imprints are erased in primordial germ cells, established in the germline according to the sex of the embryo, and maintained throughout life. Interestingly, most ICRs have oocyte derived methylation and only four imprinted domains have an ICR which is marked in the paternal germline. An interesting aspect of the 11p15/dist7 region is that the two ICRs carry opposite germline imprints, with ICR1 being marked in the male germline and ICR2 in the female germline.
Establishment of imprints
It is well established that both DNMT3A and DNMT3L methylate both ICRs in germ cells (ICR1 in sperm and ICR2 in oocytes) (reviewed in Arnaud13) (table 1 and figure 2). It is less clear how DNMTs (DNA methyltransferases) are attracted to ICR1 and ICR2 in sperm and oocytes, respectively, and how ICR1 and ICR2 are protected from DNA methylation in oocytes and sperm, respectively. Regarding ICR1, CTCFL/BORIS, a paralog of CTCF with preferential expression in male germ cells, has been proposed to direct DNA methylation at ICR1 in sperm.14 A subset of CTCFL/BORIS protein isoforms binds the human ICR1 domain.15 CTCFL/BORIS knockout mice display spermatogenesis defects but whether loss of expression of CTCFL/BORIS affects the establishment of ICR1 DNA methylation is unknown.16 It is also clear that CTCF is not required to protect ICR1 from methylation in oocytes.17–20 Nothing is known about the factors which attract methylation to ICR2 in oocytes or protect ICR2 from methylation in sperm (figure 2).
Factors involved in the establishment and/or the maintenance of 11p15/dist7 genomic imprinting

Factors involved in the establishment and the maintenance of imprinting control region (ICR)1 and ICR2 imprints of the 11p15/distal chromosome 7 region. The two ICRs with their respective DNA methylation status (black ovals for methylated and white ovals for unmethylated) are represented. Factors involved in the establishment and/or the maintenance of ICR1 or ICR2 imprints are shown with some factors of oocyte origin also involved in the maintenance of imprints in the zygote. Strikethrough factors are factors that have been shown as dispensable. This figure is only reproduced in colour in the online version.
Maintenance of imprints
Another crucial step in the imprinting cycle occurs after fertilisation when imprinted regions have to resist a wave of genome-wide demethylation followed by a wave of de novo methylation. Maternal and zygotically expressed DNMT1s are crucial to maintain the methylation imprints at both ICR1 and ICR2 on the paternal and the maternal allele, respectively. Several other factors have been implicated in the protection of the methylated paternal ICR1 allele. They include STELLA (PGC7; DPPA3) (oocyte origin),21 KAP1 (TRIM 28, TIF1β) (oocyte origin), a partner of ZFP5722 and MBD3 (zygote origin).23 CTCF17–19 and possibly the OCT4 and SOX2 pluripotency factors24 (see below) are required to protect the maternal ICR1 allele against DNA methylation. Several factors have also been implicated in the protection of the methylated maternal ICR2 allele. They include ZFP57,25 NLRP2,26 and NRLP7.27 It is likely that CTCF also plays an indirect role in the protection of the methylated maternal ICR2 allele by a mechanism involving long range chromatin looping (see below). No factors have been identified as being involved in the protection against DNA methylation of the paternal ICR2 allele. However, the Kcnq1ot1 promoter itself is required for the protection. Indeed, upon paternal transmission of a deleted promoter, ICR2 is methylated during pre-implantation development.29 Whether specific transcription factor binding sites in the promoter or the process of transcription itself account for the protection against post-fertilisation de novo methylation is unknown.
Imprinting effects of parental duplications of the 11p15/dist7 region
More than two decades ago, it was shown that two imprinting effects are associated with mouse distal chromosome 7 (Chr 7).30 Maternal duplication of distal Chr 7 (MatDup dist7) results in late embryonic lethality with embryonic and placental growth retardation, whereas paternal duplication of distal Chr 7 (PatDup dist7) results in early embryonic lethality.30 The maternal introduction of a mutant ICR1 (lacking insulator activity19) in MatDup dist7 mice corrects the growth phenotype and allows survival to adulthood, suggesting that lack of IGF2 is likely to be responsible for the growth retardation of MatDup dist7.31 PatDup dist7 mice show a phenotype similar to mice displaying knockout of the Ascl2 gene, which is essential for placenta development.32 Although the introduction of an Ascl2 transgene in PatDup dist7 mice allows survival to late gestation,32 misexpressions of other imprinted genes (including loss of CDKN1C expression and overexpression of IGF2), account for other phenotypes of PatDup dist7 mice.32
Those two duplications of distal Chr 7 mouse models are relevant to human disorders caused by dysregulation of 11p15 imprinting with MatDup dist7 and PatDup dist7 mimicking Silver-Russell syndrome (SRS) and Beckwith-Wiedemann syndrome (BWS), respectively. Approximately 10 unbalanced translocations resulting in 11p15 trisomy (involving the whole 11p15 region) have been described33–39 (table 2). They result in growth retardation or a SRS phenotype when they are maternally transmitted and overgrowth or a BWS phenotype when they are paternally transmitted (reviewed in Bliek et al,37 Demars and Gicquel,40 Begemann et al41). There are no mouse models of duplications confined to one of the two imprinted domains. However, such cis-duplications have been identified in both BWS and SRS (table 2). Cis-duplications involving the whole IGF2/H19 ICR1 domain always result in BWS if the paternal chromosome is involved, with no phenotype if the maternal chromosome is involved.3 ,35 ,37 ,42 On the other hand, cis-duplications involving the whole KCNQ1/CDKN1C ICR2 domain result in SRS if the maternal chromosome is involved with no phenotype if the paternal chromosome is involved43 ,44 (table 2). Rare cis-duplications involving only part of one of the two imprinted domains have also been identified. When they involve only part of the IGF2/H19 ICR1 domain (the ICR and the H19 gene), they result in a SRS phenotype only if maternally inherited when there is no phenotype upon paternal transmission3 ,41 (table 2). Partial cis-duplications of the CDKN1C/KCNQ1 ICR2 domain result in a BWS phenotype only if maternally inherited when there is no phenotype upon paternal transmission3 ,45 (table 2).
Duplications described in patients with Beckwith-Wiedemann syndrome (BWS) and Silver-Russell syndrome (SRS)
The IGF2/H19 ICR1 domain
Nearly a decade ago, in an extensive review, Arney46 compiled numerous genetic experiments carried out on the Igf2/H19 ICR1 domain and emphasised the complexity of the region and contradictions between some of the models, especially when it came to expression data. The addition of new mouse models which disrupt the Igf2/H19 ICR1 domain as well as the identification of new trans-regulatory factors and new genetic defects in BWS and SRS make things even more complex in 2012.
The mouse ICR1 deletion/mutation models developed between 2003 and 2011 have been initially designed to investigate the exact role of CTCF in protecting the ICR1 maternal allele from DNA methylation. Meanwhile, other trans-acting factors have been identified and increasing data have shown that CTCF function is modulated by neighbouring DNA binding factors.47 It is important to discuss the role of those other factors in order to analyse accurately the various mouse models. One of the factors is the ZFP57 KRAB zinc finger protein. ZFP57 is important for the establishment (at least at the Snrpn locus) and mostly the maintenance of DNA methylation at several maternal and paternal ICRs (table 1), although ICR1 methylation is not affected in ZFP57 deficient mice.25 ZFP57 binds all ICRs and recruits KAP1 and other chromatin modifiers (including DNMTs)48 to the parental allele carrying repressive chromatin marks (ie, paternal ICR1 and maternal ICR2 alleles).49 Moreover, oocyte depletion of KAP1 affects (although inconstantly) the maintenance of methylation only at ICR1.22 There are some ZFP57 motifs within ICR1 in both mice (six motifs) and humans (nine motifs), some of them immunoprecipitated in CHIP experiments.49 Moreover, an interesting aspect of these ZFP57 hexanucleotide (TGCCGC) binding motifs is that they overlap three of the four CTCF binding sites of the mouse ICR1 domain and the seven CTCF binding sites of the human ICR1 domain (figure 3A and B).

The IGF2/H19 imprinting control region (ICR)1 domain. (A) Mouse ICR1 (H19 DMD) with location of the various transcription factor binding sites and various deletions within ICR1. (B) Human ICR1 with location of the various transcription factor binding sites. ICR1 is organised into two clusters each containing one 450 bp A-repeat followed by three to four 400 bp B-repeats. Arrows describe hot spot deletion breakpoints within the ICR1 domain at three different highly repetitive sequences: GAGCCTGCACTGCCGCCGC GCGGCCACTT within CTCF binding sites, TGCACCCTGGGGTGAATC (grey arrows) and GAAGGGGAAGCCTCCAGAAATACACATGTGCTATGCAAGAGCCCC (black arrows). Localisation of the various deletions (plain lines) and insertion (dotted line) described in Beckwith-Wiedemann syndrome patients. Stars correspond to small deletion and point mutations within pluripotency factor binding sites. (C) Various mutations of CTCF binding sites performed in the mouse model and their consequences in CTCF binding and growth phenotype. CTCF consensus motifs are in bold, ZPF57 motifs are underlined and mutations are in lower case. This figure is only reproduced in colour in the online version.
Deletions (1.2 and 1.6 kb) involving two or three CTCF binding sites (as well as the corresponding ZFP57 motives) within the mouse ICR1 (figure 3A) result in fetal growth retardation on paternal transmission and overgrowth on maternal transmission.50 ,51 A plausible explanation to the opposite phenotypes upon different parental transmission is that CTCF binding sites are lost on maternal transmission, whereas the ZFP57 binding sites are lost on paternal transmission. However, the situation is different in humans. Indeed, whereas in humans, ZFP57 motifs also coincide with CTCF binding sites, deletions removing two to six of the seven CTCF binding sites (figure 3B) result in a BWS phenotype upon maternal transmission but do not result in any phenotype upon paternal transmission.3 ,24 ,52–56
Various studies also mutated three to four of the CTCF binding sites of the mouse ICR1.17–19 ,57 ,58 Most mutations17–19 ,57 abrogated CTCF binding resulting in loss of the insulator function and overgrowth upon maternal transmission. They did not result in any phenotype upon paternal transmission although the ZFP57 motifs were deleted in at least two of the models19 ,57 (figure 3C). The fifth model58 gives the opposite phenotype with a gain of insulator function upon paternal transmission and fetal growth retardation. In this model, the mutations (mutations of nine of the 10 CpG dinucleotides present in the four CTCF binding sites) (figure 3C) were less severe and did not affect the binding of CTCF. The absence of most CpG dinucleotides prevented the methylation on the paternal allele and therefore allowed the binding of CTCF. The possibility of mutations within CTCF binding sites has been addressed in both BWS and SRS, but such mutations do not account for BWS24 ,56 or SRS.24
Other factors possibly involved in the regulation of ICR1 imprinting are pluripotency factors. We and others recently identified mutations/small deletions of OCT4 and SOX2 binding sites within ICR1 in BWS patients with ICR1 gain of methylation (figure 3B).24 ,59 In the four cases described so far, the BWS phenotype segregated with transmission of the mutation through the female germline with no phenotype upon paternal transmission. It is also interesting to notice that all ICR1 maternal deletions described in the previous paragraph involved the OCT4 binding sites of the A2 repeat24 ,59 (figure 3B). Some OCT4 binding sites are also present within the mouse ICR160 ,61 (figure 3A) and have been shown to participate to the protection against methylation of the ICR1 maternal allele.60 OCT4 and SOX2 binding sites have also been identified at the Angelman imprinting centre.62 The mechanism of action of pluripotency factors is not clear but OCT4 was previously shown to be involved in epigenetic regulation and to interact with CTCF in regulating the X chromosome inactivation process.63 Moreover, a novel function has recently been attributed to OCT4, as a negative regulator of loop formation mediated by cohesin at CTCF binding sites.64 These OCT4 binding sites have not been specifically targeted in the mouse but have been indirectly investigated in a model with a 0.9 kb deletion of intervening sequence between the second and the third CTCF binding sites (figure 3A).65 However, maternal transmission of the deletion did not result in any phenotype or disruption of imprinting.
In summary:
-
CTCF is crucial for the protection of the maternal ICR1 allele against methylation. The only mechanisms affecting CTCF binding sites in human diseases are maternal deletions within ICR1. They result in a BWS phenotype.
-
Mutations affecting pluripotency factor binding sites result in the same phenotype and strongly suggest that in addition to CTCF, pluripotency factors play a role in the protection of the maternal ICR1 allele against methylation. Further studies will highlight their exact role in regulation of ICR1 imprinting.
-
The role of ZFP57 in the regulation of ICR1 imprinting is more questionable for several reasons: (1) deletions of ZFP57 motifs do not result in any phenotype on paternal transmission19 ,57; (2) ICR1 methylation is not affected in ZFP57 deficient mice25 although it is partially affected in ZFP57 null embryonic stem (ES) cells48; (3) patients with multilocus hypomethylation disorder caused by ZFP57 mutations do not display ICR1 loss of methylation;66 (4) no ZFP57 mutations have been identified in SRS patients with ICR1 loss of methylation.67
-
The putative role of the HOTS tumour suppressor in the pathogenesis of BWS/SRS has to be investigated.8
The CDKN1C/KCNQ1 ICR2 domain
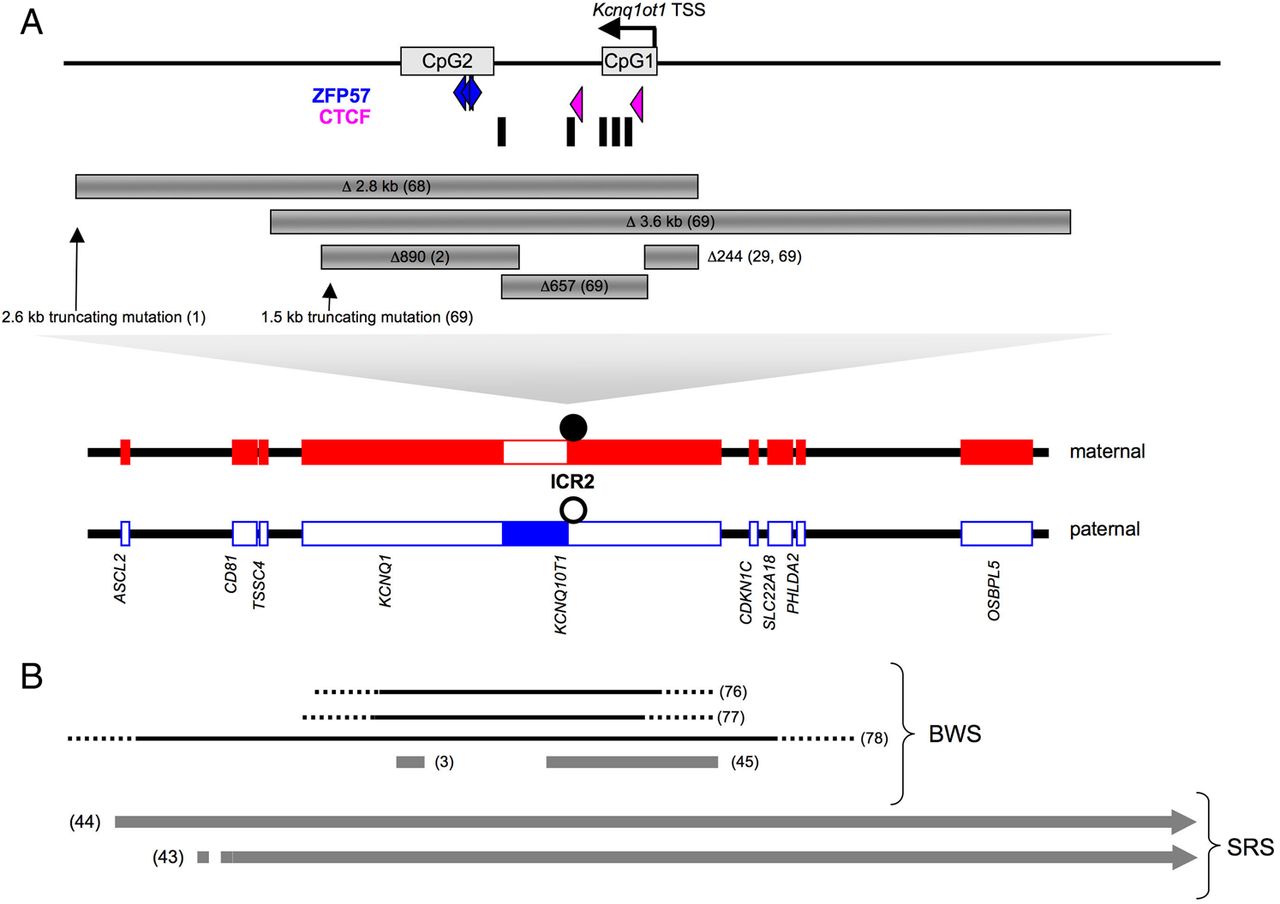
The crucial role of ICR2 and the Kcnq1ot1 ncRNA is highlighted by several mouse models deleting the DMR,68 ,69 the Kcnq1ot1 promoter29 ,69 or truncating the Kcnq1ot1 RNA1 ,69 (figure 4A). Upon paternal transmission, those defects result in fetal growth retardation and relaxation of imprinting with re-expression of normally silent paternal genes. It is very likely that the growth restriction phenotype is caused by the re-expression of the paternal Cdkn1c allele.68 ,69 Upon maternal transmission, there is no growth phenotype and the imprinting is not affected. The truncating mutation models1 ,69 suggest that control of ICR2 imprinting is achieved through the transcription of the full Kcnq1ot1 RNA as premature termination of Kcnq1ot1 results in re-expression of normally silent paternal genes. However, one of the truncating mutation models (truncation at 2.6 kb of the Kcnq1ot1 transcription start site)1 (figure 4A) suggests that an additional mechanism is at work. Indeed, this mutation affects the imprinting status of Cdkn1c in a tissue specific manner with retention of monoallelic Cdkn1c expression in some tissues such as kidney or liver.1 Moreover, the mechanism involving transcription of a full Kcnq1ot1 RNA has recently been challenged by Kcnq1ot1 RNA depletion. Kcnq1ot1 RNA depletion (by more than 80%) in stem cells does not affect imprinted gene expression, favouring a mechanism by which the act of transcription rather that the transcript itself regulates imprint maintenance.10

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The CDKN1C/KCNQ1OT1 domain. (A) Mouse imprinting control region (ICR)2 domain with the location of ZFP57 and CTCF binding sites, MD1 repeats (black boxes) and CpG islands (CpG1 and CpG2). The various deletions and truncating mutations are also shown. (B) Human ICR2 domain with the deletions (black) and the cis-duplications (grey) described in Beckwith-Wiedemann and Silver-Russell syndromes. This figure is only reproduced in colour in the online version.
In humans, abnormal imprinting of the CDKN1C/KCNQ1 domain is very common in BWS. Indeed, since the first description of the defect more than a decade ago,70 ,71 it is well established that approximately 60% of BWS patients display loss of DNA methylation at ICR2 (reviewed in Demars and Gicquel40). The ICR2 loss of methylation on the maternal allele is associated with biallelic expression of KCNQ1OT1 and loss of expression of genes normally expressed from the maternal allele, including CDKN1C. For a quarter of those patients, the mechanism involves the dysregulation of trans-acting factors as the loss of DNA methylation affects imprinted loci other than 11p15 ICR2.72–75 For the remaining 75% of patients, a mechanism in cis is rarely identified.3 ,76 Two deletions within KCNQ1 have been described in humans76 ,77 (figure 4B). Although their boundaries have not been precisely identified, those two 255–397 kb deletions encompass ICR2, as well as the whole KCNQ1OT1 transcript, but respect the CDKN1C gene which in both cases is downregulated (figure 4B). A larger third deletion covering at least 860 kb and including the CDKN1C gene also results in BWS phenotype upon maternal transmission78 (figure 4B).
When deletions of ICR2 or the Kcnq1ot1 promoter in the mouse result in growth retardation upon paternal transmission, human deletions give a BWS phenotype upon maternal transmission. The deletions described in humans are much larger than mouse deletions (figure 4A and B) and a likely explanation for these discrepancies is the existence of regulatory elements other than ICR2 and KCNQ1OT1 in the imprinting domain (see below). Deletions3 ,76 as well as other copy number variants3 are very rare in BWS with ICR2 loss of methylation. However, some recently described small cis-duplications3 ,45 strongly suggest the existence of cis-regulatory elements other than ICR2 and KCNQ1OT1. We recently described a small 50 kb cis-duplication within the KCNQ1 gene which does not involve ICR2 or KCNQ1OT1.3 This cis-duplication encompasses a region with strong binding sites for CTCF and cohesin (RAD21 subunit) within the second intron of the KCNQ1 gene. As discussed previously, CTCF and cohesin have been shown to mediate intrachromosomal looping interactions at the IGF2/H19 ICR1 domain.4 ,79 We therefore hypothesise that the duplication of this 50 kb KCNQ1 region impairs chromatin looping. This hypothesis is reinforced by the very recent description in mouse ES cells of a 314 kb intrachromosomal repressive loop connecting a CTCF binding site located in the second intron of KCNQ1 to a CTCF binding site 3′ of CDKN1C, therefore encompassing ICR2.80 It is noteworthy that the three human deletions previously described76–78 encompass this putative cis-regulatory region, which is likely to be important for the maintenance of ICR2 DNA methylation (figure 4B).
Whereas CDKN1C plays a crucial role in fetal growth,81 ,82 the CDKN1C/KCNQ1 domain is exceptionally involved in SRS. Two cis-duplications involving the whole CDKN1C/KCNQ1 domain43 ,44 (figure 4B) have been described and they result in a SRS phenotype upon maternal transmission with no phenotype upon paternal transmission. The growth phenotype is explained by the increase of CDKN1C expression upon maternal transmission when its expression is unchanged on paternal transmission. The ICR2 methylation status has been extensively studied in SRS and SRS-like patients and has always been reported as normal. However, because Cdkn1c transgenic mice display a SRS phenotype81 and because some deletions2 and truncating mutations of Kcnq1ot11 ,69 result in increased Cdkn1c expression without abnormal ICR2 methylation defect, it might be interesting to investigate such a mechanism in SRS patients. This hypothesis is strengthened by the recent description of activating mutations of CDKN1C in the IMAGe (intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, genital abnormalities) syndrome which shares some phenotypes with SRS, such as fetal growth retardation or facial dysmorphia.83
The exact role of ZFP57 in ICR2 imprinting is not completely understood. Several studies in mice and humans suggest a role for this factor in the maintenance of methylation at the maternal ICR2. (1) ICR2 is partially demethylated in approximately one third of patients displaying ZFP57 mutations.66 On the other hand, ZFP57 mutations have not been identified in BWS patients with isolated ICR2 loss of methylation or their mothers.84 (2) ZFP57 binds the maternal ICR2 in murine ES cells and ICR2 is completely demethylated in ZFP57 null ES cells.49 However, whether ZFP57 is involved in the establishment of ICR2 methylation in the oocyte is unknown, as the methylation status of ICR2 was not analysed in ZFP57 deficient mice.25 There are some ZFP57 binding sites in the mouse and human ICR2 (figure 4A). It would be interesting to see whether mutations of these binding sites in the mouse result in ICR2 loss of imprinting and whether mutations of these ZFP57 binding sites on the maternal allele of ICR2 account for some BWS cases.
In summary:
-
The discordances between the mouse and human ICR2 deletion models regarding the growth phenotype and the pattern of parental transmission are probably explained by the existence of other cis-regulatory elements at distance of ICR2 and KCNQ1OT1 which are affected in human deletions (sizes between 255–850 kb) but not in mouse models where the larger deletion is 3.6 kb (figure 4). Little is known about the centromeric 11p15 imprinted region apart from ICR2 itself. Our recent findings suggest that a region located in the second intron of KCNQ1 and harbouring strong CTCF and cohesin binding sites acts as a cis-regulatory element for genomic imprinting of the CDKN1C/KCNQ1 region. Genetic manipulation of this region in the mouse will ultimately answer this question.
-
More studies are needed to establish precisely the role of ZFP57 in the regulation of genomic imprinting of the CDKN1C/KCNQ1 domain and its involvement in the pathogenesis of BWS.
-
Given the role of the CDKN1C gene in the regulation of fetal growth, more studies are needed to investigate the putative role of the CDKN1C/KCNQ1 domain in the pathogenesis of SRS.
Conclusion
Although the two imprinted domains of the 11p15/dist7 region are very similar in humans and mice, there are discrepancies in phenotypes and the pattern of parental transmission between genetically engineered mouse models and human genetic defects. We therefore aimed at revisiting those various models, in light of recent knowledge regarding the regulation of genomic imprinting of the 11p15/dist7 region and recent description of novel molecular defects in patients with SRS or BWS. Although rare, these recently described molecular defects provide highly valuable clues in terms of the mechanisms that establish and maintain proper imprinting at this crucial fetal growth regulatory region.
The essential messages are the following: (1) In addition to ICR2, a region located within the second intron of KCNQ1 is likely to act as a cis-regulatory element and to play a role in the protection of ICR2 DNA methylation on the maternal allele. This is probably achieved through a large CTCF/cohesin mediated chromatin loop insulating the methylated maternal ICR2. CTCF is therefore likely to play a role within the CDKN1C/KCNQ1 domain as important as it does within the IGF2/H19 domain. Mouse models deleted in this region will definitively be crucial to confirm the exact role of this new cis-regulatory element. (2) There are increasing data showing that CTCF function is modulated by neighbouring DNA binding factors. The recent description of mutations of pluripotency factor binding sites in BWS patients makes pluripotency factors excellent candidates for either prevention of methylation of the maternal ICR1 in oocytes or maintenance of the unmethylated state after fertilisation. The description of those unexpected mutations will allow to turn to the mouse model to specifically mutate these pluripotency factor binding sites. (3) More clinical studies are needed to appreciate clearly how the ZFP57/KAP1 complex can result in 11p15 imprinting disorders. One aspect of clinical research in this field should look at mutation of the ZFP57 binding site within ICR1 and ICR2 in SRS and BWS, respectively.
References
Footnotes
-
Contributors Both authors contributed to the design and writing of this manuscript.
-
Funding None.
-
Competing interests This work was supported by the National Health and Medical Research Council of Australia (Project grant 472637), the Baker IDI Heart and Diabetes Institute and the Victorian Government’s OIS Program
-
Provenance and peer review Not commissioned; externally peer reviewed.