Article Text

Abstract
Background Male infertility is a prevalent issue worldwide, mostly due to the impaired sperm motility. Multiple morphological abnormalities of the sperm flagella (MMAF) present aberrant spermatozoa with absent, short, coiled, bent and irregular-calibre flagella resulting in severely decreased motility. Previous studies reported several MMAF-associated genes accounting for approximately half of MMAF cases.
Methods and result We conducted genetic analysis using whole-exome sequencing in 88 Han Chinese MMAF probands. CFAP65 homozygous mutations were identified in four unrelated consanguineous families, and CFAP65 compound heterozygous mutations were found in two unrelated cases with MMAF. All these CFAP65 mutations were null, including four frameshift mutations (c.1775delC [p.Pro592Leufs*8], c.3072_3079dup [p.Arg1027Profs*41], c.1946delC [p.Pro649Argfs*5] and c.1580delT [p.Leu527Argfs*31]) and three stop-gain mutations (c.4855C>T [p.Arg1619*], c.5270T>A [p.Leu1757*] and c.5341G>T [p.Glu1781*]). Additionally, two homozygous CFAP65 variants likely affecting splicing were identified in two MMAF-affected men of Tunisian and Iranian ancestries, respectively. These biallelic variants of CFAP65 were verified by Sanger sequencing and were absent or very rare in large data sets aggregating sequence information from various human populations. CFAP65, encoding the cilia and flagella associated protein 65, is highly and preferentially expressed in the testis. Here we also generated a frameshift mutation in mouse orthologue Cfap65 using CRISPR-Cas9 technology. Remarkably, the phenotypes of Cfap65-mutated male mice were consistent with human MMAF.
Conclusions Our experimental observations performed on both human subjects and on Cfap65-mutated mice demonstrate that the presence of biallelic mutations in CFAP65 causes the MMAF phenotype and impairs sperm motility.
- male infertility
- exome sequencing
- mouse model
- sperm flagella
- CFAP65
Statistics from Altmetric.com
Introduction
Male infertility is a common reproduction defect, affecting approximately 15% of couples worldwide.1 Sperm flagellar formation occurs at the end of spermatogenesis, concurrently with head compaction and reshaping.2–4 Disorders of sperm flagella impair sperm locomotion, which is indispensable for natural fertilisation. Over 80% cases with male infertility present some alteration of sperm motility.5 The multiple morphological abnormalities of the sperm flagella (MMAF) phenotypes are characterised by five concomitant typical aberrant morphologies of the flagella, including absent, short, coiled, bent and/or irregular-calibre flagella, associated with dramatically declined motility.6
Previous genetic studies revealed that genetic factors are the major causes of human MMAF syndrome,7 and biallelic mutations in DNAH1 were first identified to be causes of the defect.6 8 Subsequently, cilia and flagella associated proteins (CFAPs), including CFAP43, CFAP44,9 10 CFAP6911 12 and CFAP25113–15 were demonstrated to be a recurrent cause of MMAF. Some other genes have also been recently reported to be causative to MMAF, including QRICH2,16 FSIP2,17 18 ARMC2,19 SPEF2 20 and TTC21A.21 However, Coutton et al 19 noted only 32.3% diagnostic efficiency in 167 individuals suffering from MMAF, confirming the important genetic heterogeneity of MMAF and highlighting the fact that many more pathological genes remain to be identified for this defect. The genetic causes and pathogenic mechanisms in remaining unresolved MMAF cases still need to be elucidated.
Our previous study identified a potential pathogenic homozygous mutation of CFAP65 (also known as coiled-coil domain containing (CCDC) CCDC108 (MIM: 614 270)) in one MMAF proband.9 Here we recruited 88 additional Han Chinese probands with MMAF from two centres in China. Through genetic analysis using whole-exome sequencing (WES), we identified six probands with biallelic null mutations in CFAP65 from unrelated families, accounting for 6.8% (6/88) of our MMAF cohort. In addition, analysis of the previously described cohort of 167 subjects of mixed origin19 permitted to identify two additional patients with likely deleterious homozygous CFAP65 variants. We observed that CFAP65, encoding CFAP65, is highly and preferentially expressed in testis. We also generated an animal model for mouse orthologue Cfap65. Notably, Cfap65-mutated male mice manifest the MMAF phenotypes. Our findings further confirm that CFAP65 biallelic mutations induce a typical MMAF phenotype with a severely impaired sperm motility and primary male infertility.
Materials and methods
Probands participants
All 88 MMAF cases were enrolled at the First Affiliated Hospital of Anhui Medical University and West China Second University Hospital in China. They came from unrelated families and were diagnosed with primary infertility according to their inability to conceive spontaneously after at least 1 year of natural attempts in the absence of contraceptive measures. Parental consanguinity was self-reported by the probands in four families (subjects A005 II-1, A037 II-1, A052 II-1 and H010 II-1). No other reproduction problems such as ejaculation disorder, erectile dysfunction and orchiatrophy were detected. The chromosomal karyotypes of these cases were normal (46, XY), and there was no deletion detected in the human Y chromosome. In addition, primary ciliary dyskinesia (PCD) related symptoms such as sinusitis, bronchitis, pneumonia and otitis media were also excluded. Detailed semen analyses showed the dramatic decrease of sperm motility. Peripheral whole blood samples from patients and their available parents were collected for genetic analyses.
In addition, we analysed the data obtained by WES performed for a total of 167 individuals affected by primary infertility associated with an MMAF syndrome previously described by a French team from Grenoble, France.19 All recruited individuals displayed isolated infertility with no other clinical features; in particular, PCD syndrome was excluded. In this cohort, 83 individuals originated from North Africa and sought consultation for primary infertility at the ‘Clinique des Jasmins’ in Tunis, 52 individuals originated from the Middle East (Iran) and were treated in Tehran at the Royan Institute (Reproductive Biomedicine Research Centre) for primary infertility and 32 individuals were recruited in France, mainly at the Cochin Institute in Paris. All individuals presented with a typical MMAF phenotype. All individuals had a normal somatic karyotype (46, XY) with normal bilateral testicular size, hormone levels and secondary sexual characteristics.
WES and data analyses
Genomic DNA was isolated from peripheral blood of MMAF patients using DNeasy Blood and Tissue kit (QIAGEN). The whole exomes of patients were extracted and enriched following the protocol of Agilent SureSelectXT Human All Exon Kit. Illumina HiSeq X-TEN platform was employed for DNA sequencing. The follow-up bioinformatic study was conducted for standard assembly (Burrows-Wheeler Aligner), calling (Genome analysis Toolkit) and annotation (ANNOVAR). The details of analyses have been described previously.9 13 Data from the 167 strong cohort were analysed as described by Coutton et al.19
Semen characteristics and sperm morphological study
Fresh semen samples of recruited patients were collected by masturbation after 2‒7 days of sexual abstinence and examined after liquefaction for 30 min at 37°C. Semen volume, sperm concentration, sperm progressive motility and motility were accessed following the WHO guidelines. Fresh semen samples of Cfap65-mutated male mice were collected from single cauda epididymis and diluted in 1 mL sperm rinse (10101, Vitrolife). The semen samples were examined after incubation for 10 min at 37°C by computer-assisted analysis system (Cyto-S, VideoTesT). Over four 8-week-old Cfap65-mutated and wild-type male mice were measured. The remaining of human and mouse semen samples were fixed in 4% paraformaldehyde over 24 hours respectively and subsequently spread on the slides. The H&E staining was conducted in standard methods for sperm morphology study. Over 200 spermatozoa were examined, and morphological abnormalities were evaluated following the WHO guidelines. One spermatozoon was classified into only one major morphological category.22
Generation of Cfap65-mutated mouse model
We used the CRISPR/Cas9 technology to generate Cfap65-mutated mice. The single-guide RNA (sgRNA) was synthesised and ligated to the pX458 plasmid, digested with BbsI before. Single colonies were picked and verified by Sanger sequencing. C57BL/6 wild-type female mice were superovulated and mated with wild-type male mice to obtain zygotes. The plasmid harbouring sgRNA was purified and injected into the cytoplasm of zygotes. After in vitro culture to blastocysts, they were transferred into pseudopregnant female mice. The founder mice with Cfap65 frameshift mutations were mated with wild-type mice. The genotyping of their offspring was verified by PCR and Sanger sequencing. This study was carried out in accordance with the recommendation of the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health.
Transmission electron microscopy (TEM) evaluation
Fresh samples of human semen and mouse testis tissues were fixed in 2.5% phosphate buffered glutaraldehyde. After dehydration and infiltration, samples were embedded in Epon 812, and ultrathin sections were stained with uranyl acetate and lead citrate. The ultrastructures were observed and recorded by TEM (TECHAI-10, Philips) with an accelerating voltage of 80 kV.
TUNEL analysis
Mouse testes were collected and fixed in modified Davidson’s fluid for up to 24 hours and stored in 70% ethanol. Then the samples were dehydrated through a graded ethanol series and embedded in paraffin. Tissue sections were prepared and mounted on glass slides. Apoptotic cells in testis were detected using the terminal deoxynucleotidyl transferase dUTP nick end-labelling (TUNEL) assay (A113, Vazyme) according to the manufacturer’s instructions.
Results
Comparison of the WES data with the human reference genome (hg19) permitted to identify approximately 120 000‒140 000 single nucleotide variants and indels per proband. Due to high intolerance of infertility under natural selection pressure, our analysis emphasised on rare variants with allele frequencies <1% in large genome data sets from the 1000 Genomes Project and Genome Aggregation Database (gnomAD). We gave priorities to null variants (such as stop-gain, stop-loss, frameshift and splice-site mutations). Damaging missense variants predicted by MutationTaster, SIFT and PolyPhen-2 were also taken into consideration. Genetic defects so far identified to be responsible for MMAF syndrome have always been shown to segregate under recessive transmission. Therefore, homozygous and potential compound heterozygous mutations were investigated.6 9 Moreover, the mutations in the genes with specific or preferential expressions in testis or those implicated in flagellar function were preferred.
Interestingly, we found six Chinese patients carrying biallelic null mutations in CFAP65 in this study (figure 1 and online supplementary figure 1). The homozygous frameshift mutations c.1775delC (p.Pro592Leufs*8) and c.1580delT (p.Leu527Argfs*31) were identified in subjects A005 II-1 and A037 II-1, respectively. We also found homozygous stop-gain mutations c.5341G >T (p.Glu1781*) and c.4855C >T (p.Arg1619*) in subjects A065 II-2 and H010 II-1, respectively. In addition to these homozygous variants, we identified two additional compound heterozygotes with CFAP65 null variants: subject A018 II-1 carried c.3072_3079dupCTATTACC (p.Arg1027Profs*41) and c.1946delC (p.Pro649Argfs*5), and subject A065 II-2 carried c.4855C >T (p.Arg1619*) and c.5270T >A (p.Leu1757*).
Supplemental material

Pedigrees of six Chinese MMAF probands with biallelic null mutations of CFAP65. Biallelic CFAP65 mutations were verified by Sanger sequencing, and results obtained from the relatives available were shown. The biallelic mutations in subject A018 II-1 were also confirmed through picking single colonies with the regions containing both mutations. Black arrows showed the direction of Sanger sequencing. Red arrows indicate the positions of point mutations, and red rectangles displayed frameshift sequences in deletion or duplication mutations. MMAF, multiple morphological abnormalities of the sperm flagella; WT, wild type.
All these CFAP65 variants were verified by Sanger sequencing in the probands and their available relatives (figure 1). Clone sequencing was employed for haplotyping and verification of biallelic CFAP65 mutations in subject A018 II-1 (figure 1). Notably, all these CFAP65 null mutations were absent or very rare in the human population genome data archived in the 1000 Genomes Project and gnomAD databases (table 1). CFAP65 is highly and specifically expressed in the testis according to the ENCODE, FANTOM and GTEx databases. These findings further suggested that biallelic null mutations of CFAP65 could be pathogenic in human MMAF.
Biallelic CFAP65 mutations identified in MMAF probands
Exome data from the French cohort were reanalysed. Previous analyses of this cohort permitted to identify homozygous variants in a total of 60 men (36%) in confirmed MMAF-associated genes leaving 107 undiagnosed individuals. In these undiagnosed individuals, two carried likely deleterious CFAP65 homozygous variants (table 1 and online supplementary figure 1). One patient was Tunisian (17IF078) and carried the CFAP65 variant c.3047T >G (p.Leu1016Arg). This variant is predicted by HSF (Human Splicing Finder, http://www.umd.be/HSF3/) to activate an exonic cryptic donor site likely altering splicing and is absent from the gnomAD database. Moreover, the amino acid change is predicted by SIFT to be deleterious (score of 0) and by PolyPhen-2 to be probably damaging (score of 0.98). The other CFAP65-mutated patient was of Iranian origin (17IF011) and carried the c.645G >A variant, which affects the last nucleotide of CFAP65 exon 6. This mutation located at a critical splicing site is described by HSF to alter the donor site most probably affecting CFAP65 splicing. Unfortunately, additional biological material (sperm) could not be obtained from these two subjects to perform functional assays and confirm the deleterious effect of these variants on splicing.
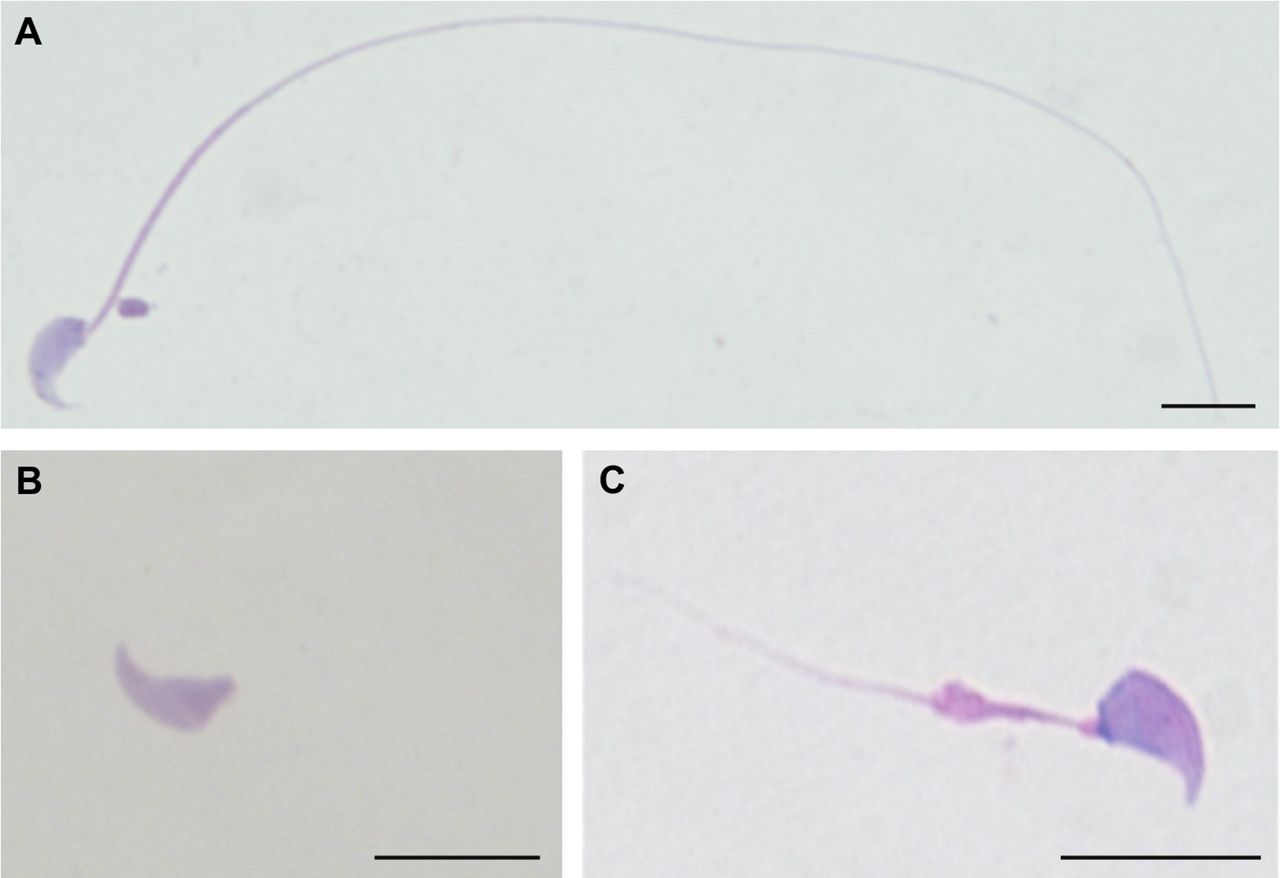
The detailed MMAF phenotypes of the six Chinese patients with biallelic null mutations of CFAP65 were ascertained. Semen analysis showed the sperm samples of all six patients totally lost the progressive motility (see online supplementary table 1). Morphological study by H&E staining illustrated the severely reduced motility resulted from sperm morphological abnormalities, including absent, short, coiled, bent and irregular-calibre flagella (figure 2). No morphologically normal spermatozoa were observed in subjects A037 II-1, A052 II-1 and H010 II-1. Sperm flagellar formation was also seriously altered in the remaining cases. Absent and short flagella were most frequently observed, accounting for >80% of the spermatozoa from the CFAP65-mutated patients. TEM further revealed the flagellar ultrastructure defects in spermatozoa of CFAP65-mutated men (figure 3A–C). Over 50 flagellar cross-sections were investigated for each CFAP65-mutated proband to access microtubule assembly in sperm flagella. The central pair of microtubules were absent in most flagella, in contrast of normal ‘9+2’ microtubule structure (nine peripheral microtubule doublets and the central pair of microtubules) in controls. Cross sections also showed hypertrophy and hyperplasia of fibrous sheaths (figure 3).

Sperm morphological study in the MMAF-affected men with biallelic null mutations of CFAP65. (A) A spermatozoon with normal morphology from a healthy man was shown under light microscopy. (B and C) The spermatozoa of the CFAP65-mutated men presented multiple flagellar malformations, of which the major morphological abnormalities are absent and short flagella. Scale bars: 10 µm. MMAF, multiple morphological abnormalities of the sperm flagella.
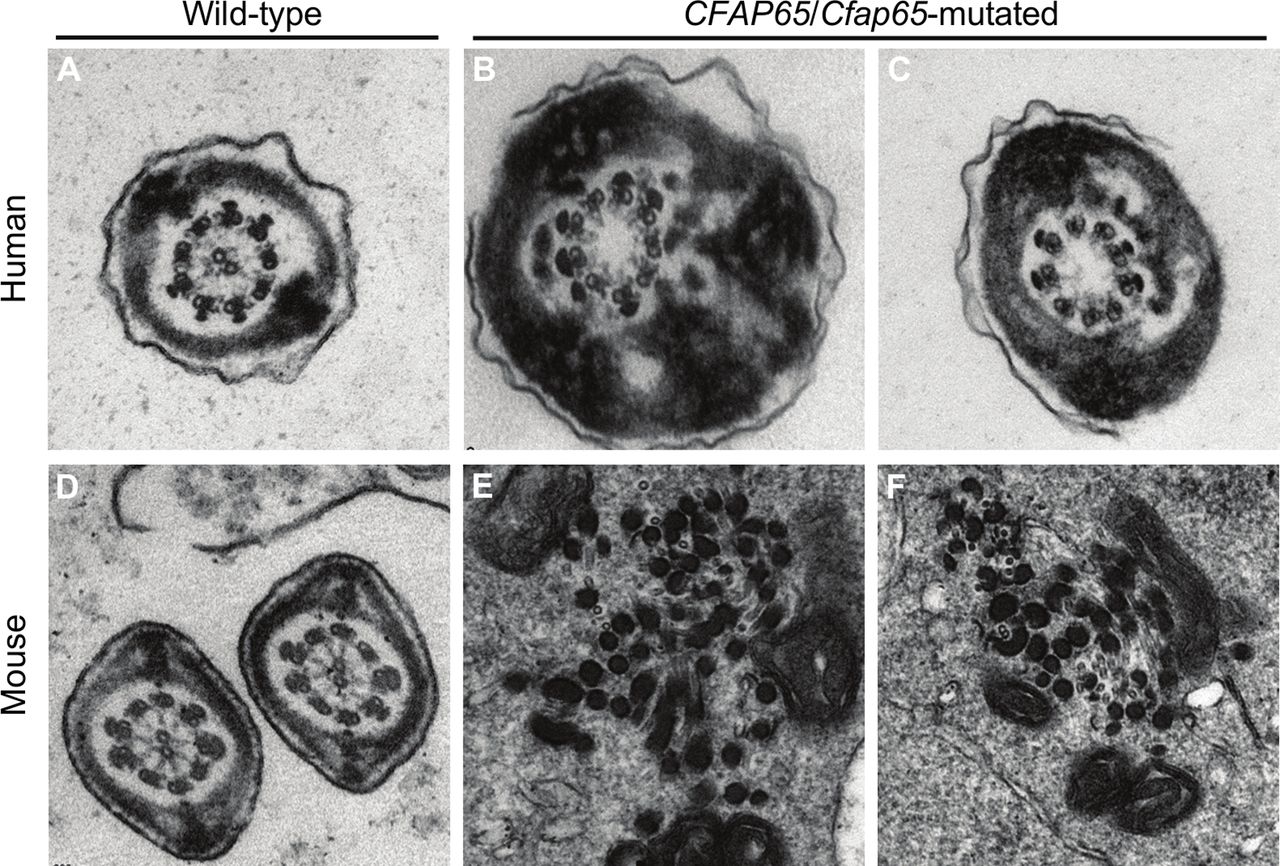
Sperm ultrastructure in CFAP65-mutated men and Cfap65-mutated mice by TEM. (A) A cross-section ultrastructure of a normal spermatozoon from a fertile man. It clearly displayed a typical ‘9+2’ microtubule structure with nine peripheral microtubule doublets and the central pair of microtubules. (B and C) Most cross-sections of spermatozoa from CFAP65-mutated subjects presented a ‘9+0’ microtubule structure lacking the central pair of microtubules. Hypertrophy and hyperplasia of fibrous sheaths were also observed. (D) The typical cross-section of ‘9+2’ microtubule structure in a normal testicular spermatozoon from a wild-type male mouse. (E and F) A completely disorganised axoneme was observed in a testicular spermatozoon from Cfap65-mutated male mice. Central microtubules were missing, and peripheral microtubule doublets were disarranged. TEM, transmission electron microscopy.
To further investigate the roles of CFAP65 in sperm flagellar formation, we generated a frameshift mutation in mouse orthologue Cfap65 using CRISPR/Cas9 technology. Sanger sequencing confirmed a 2 bp deletion in the coding region of Cfap65 (see online supplementary figure 2). Fertility was tested by breeding more than four respective male and female Cfap65-mutated mice with wild-type mice. All these male Cfap65-mutated mice had no offspring, while female Cfap65-mutated mice could give births, suggesting that the biallelic Cfap65 mutation could cause male infertility in mice.
Detailed phenotypes of Cfap65-mutated male mice were analysed. There was no significant difference in gross testis weight between Cfap65-mutated and wild-type male mice. However, semen analysis showed the complete loss of progressive motility in male Cfap65-mutated male mice (table 2). Under light microscopy, Cfap65-mutated male mice displayed typical MMAF phenotypes with severe morphological abnormalities of the sperm flagella in male (figure 4). The majority of spermatozoa displayed absent or short flagella, which is consistent with the findings in CFAP65-mutated men. No normal sperm flagella could be observed in all homozygous Cfap65-mutated male mice. Ultrastructure of mouse flagellar cross-section also showed the lack of central pair of microtubules (figure 3D–F). Moreover, the nine peripheral microtubule doublets in Cfap65-mutated male mice were scattered irregularly, highlighting a severer defect than what was observed in CFAP65-mutated men. The phenotype of Cfap65-mutated male mice was therefore highly consistent with that of CFAP65-mutated men.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sperm morphology study in Cfap65-mutated male mice. (A) Light microscopy showed a spermatozoon with normal morphology from a wild-type male mouse. (B and C) All spermatozoa from Cfap65-mutated male mice manifested aberrant flagellar morphologies, of which the major malformations are absent and short flagella, consistent with the clinical phenotypes of CFAP65-mutated men. Scale bars: 10 µm.
Semen characteristics and sperm morphology of CFAP65-mutated mice
Discussion
Efficient sperm motility is indispensable for normal fertilisation. It can be altered by aberrant shaping of sperm flagella frequently reported in infertile men. Recently, novel genetic factors have been revealed to induce MMAF, a severe form of asthenozoospermia. However, approximately half of human MMAF cases remain undiagnosed, suggesting that numerous other genes are involved in the pathogenesis of MMAF.19 Although one patient carrying a CFAP65 homozygous stop-gain mutation was identified in our previous study,9 this single case evidence cannot readily confirm the pathogenic roles of CFAP65 mutations in human MMAF. In this study, we employed WES for genetic analyses in a larger cohort of 88 Chinese MMAF subjects. Notably, we identified six MMAF-affected men carrying biallelic null mutations of CFAP65, accounting for 6.8% MMAF probands. All of these CFAP65 mutations are absent or very rare in human population genome data sets. In addition, we identified two additional MMAF-affected men originating from Iran and North Africa carrying very likely deleterious homozygous variants, thus confirming that alterations of the CFAP65 gene can affect MMAF patients irrespectively of their origins. Overall, 45 out of 88 (51.1%) Han Chinese men with MMAF carry biallelic mutations of known MMAF-associated genes, including the newly identified CFAP65 in this study (see online supplementary table 2).
Our mouse model with a Cfap65 homozygous frameshift mutation also manifested a typical MMAF phenotype, which is consistent with our findings in human patients carrying biallelic null mutations in CFAP65. Therefore, our genetic and phenotypic analyses performed on humans and mice confirm the indispensable roles of CFAP65 in flagellar formation, and formally demonstrate that the absence of CFAP65 protein leads to the MMAF phenotype in both species.
Cilia and flagella are highly conserved throughout evolution and play vital roles in many organisms. FAPs (flagellar associated proteins), orthologues of CFAPs, were identified in purified flagella from Chlamydomonas reinhardtii by mass spectrometry.23 Many human CFAPs were proven to be associated with cilia and flagella dysfunction. Lack of CFAP43, CFAP44, CFAP69 and CFAP251 (also known as FAP43, FAP44, FAP69 and FAP251, respectively) were shown to alter flagellar assembly and to be a recurrent cause of MMAF. CCDC39 (also known as FAP59) and CCDC65 (also known as FAP250) containing coiled-coil domains are also required in the assembly of inner dynein arms, and their deficiency results in PCD, a severe ciliopathy phenotype.24–26 NPHP3 (also known as FAP22) is also essential for cilia development, and its dysfunction causes polycystic kidney disease and renal-hepatic-pancreatic dysplasia.27 28 CFAP65 could also be detected in C. reinhardtii flagella, suggesting its potential function in ciliary and flagellar formation.23
CFAP65 is evolutionarily conserved in several species; however, its function has not been characterised so far.23 CFAP65 is highly and specifically expressed in the testis and has been identified in centrioles of bovine sperm and in human spermatozoa by proteomic analyses.29 30 Furthermore, a genetic variant of CFAP65 in chicken was shown to impair sperm motility in homozygous males.31 CFAP65 in both humans and mice has two ASH domains that are frequently found in proteins associated with cilia, flagella, the centrosome and the Golgi complex.32 The murine testicular transcriptome showed that only adult male mice could express abundant CFAP65 rather than juvenile mice.33 All evidence suggest the indispensable function of CFAP65 in spermatogenesis after sexual maturity. To confirm the pathogenicity of CFAP65 mutations, we generated a mouse model with 2 bp frameshift mutation in orthologue Cfap65. All Cfap65-mutated male mice were infertile, and their phenotypes were consistent with those of CFAP65-mutated men, indicating the essential roles of CFAP65 in axonemal organisation and flagellar formation. Further pathogenic mechanisms of CFAP65 mutations in MMAF still need to be illustrated in the future.
In this study, we found the phenotypic variance of sperm concentration in the CFAP65-mutated men, from the normal concentration (50.0 million/mL in subject 17IF011) to severe oligozoospermia (0.5 million/mL in subject A062 II-2) (online supplementary table 1). The majority of CFAP65-mutated men showed the sperm concentrations ranging from 4.4 to 14.5 million/mL, suggesting mild oligozoospermia. This CFAP65-associated variance in sperm concentration is similar to the previous findings in the DNAH1 and CFAP69 studies, which could result from environment factors, endocrinal level or other factors.6 11
Furthermore, the sperm concentrations of Cfap65-mutated male mice were significantly lower than those in the wild-type male mice (table 2). This observation suggested that Cfap65 deficiency could cause more severe phenotypes of oligozoospermia in mice than in humans. Here we performed TUNEL assay, which showed that the aberrant spermatozoa induced apoptosis and were phagocytised by the Sertoli cells in Cfap65-mutated male mice (see online supplementary figure 3). This phenomenon could be related to the sperm concentration decline in Cfap65-mutated male mice. Similar findings of oligozoospermia were also reported in previous studies on the sperm flagellar malformations associated with CFAP43, CFAP44 and CFAP69. 9 11
Some members of the CCDC family were reported to result in PCD.24 25 Intriguingly, CFAP65 is also known as CCDC108. In order to further exclude or investigate the potential PCD-related symptoms, we successfully recalled the cases A018 II-1 and A037 II-1. No obvious lung defects were found in either of the two cases (see online supplementary figure 4). Furthermore, we investigated the airway cilia of our Cfap65-mutated mouse model by staining the bronchial tube cross-section. Notably, the cilia and ciliated cells can be remarkably stained in both wild-type and Cfap65-mutated mice (see online supplementary figure 5).
In conclusion, our experimental observations in humans of different origins and a mouse model illustrated that biallelic mutations of CFAP65 cause MMAF and primary male infertility. We also observed that CFAP65 plays a critical role in flagellar formation and sperm motility.
Acknowledgments
We would like to thank the families for participating and supporting this study.
References
Footnotes
WeL, HW and FL contributed equally.
Contributors WeL, XH, YunC and FZ designed the study. HW, FL, XN, MingrL, QT, YS, AA-Y, JingZ, YZ, HC, YW, JW, YG, YCh, XZ, XH and YunC provided patients’ data and performed clinical assessments. WeL, ST, Z-EK, JintZ, CL, CC and WaL conducted experiments. WeL, HW, FL, LJ, MingxL, XH, PFR, YunC and FZ analysed data. WeL, HW, XH, PFR, YunC and FZ wrote the manuscript. PFR, YunC and FZ supervised the study.
Funding This work was supported by National Natural Science Foundation of China (31625015, 31521003 and 81601340), Special Foundation for Development of Science and Technology of Anhui Province (2017070802D150 and YDZX20183400004194), Foundation of the Education Department of Anhui Province (KJ2016A370), Natural Science Foundation of Anhui Province (1708085QC59 and 1908085QH313), Shanghai Medical Center of Key Programs for Female Reproductive Diseases (2017ZZ01016) and Shanghai Municipal Science and Technology Major Project (2017SHZDZX01).
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval This study was approved by the ethical committees of the centers participating in this study and the animal ethics committee at the School of Life Science, Fudan University.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.