Article Text

Abstract
Background: The vast majority of BRCA1 missense sequence variants remain uncharacterised for their possible effect on protein expression and function, and therefore are unclassified in terms of their pathogenicity. BRCA1 plays diverse cellular roles and it is unlikely that any single functional assay will accurately reflect the total cellular implications of missense mutations in this gene.
Objective: To elucidate the effect of two BRCA1 variants, 5236G>C (G1706A) and 5242C>A (A1708E) on BRCA1 function, and to survey the relative usefulness of several assays to direct the characterisation of other unclassified variants in BRCA genes.
Methods and Results: Data from a range of bioinformatic, genetic, and histopathological analyses, and in vitro functional assays indicated that the 1708E variant was associated with the disruption of different cellular functions of BRCA1. In transient transfection experiments in T47D and 293T cells, the 1708E product was mislocalised to the cytoplasm and induced centrosome amplification in 293T cells. The 1708E variant also failed to transactivate transcription of reporter constructs in mammalian transcriptional transactivation assays. In contrast, the 1706A variant displayed a phenotype comparable to wildtype BRCA1 in these assays. Consistent with functional data, tumours from 1708E carriers showed typical BRCA1 pathology, while tumour material from 1706A carriers displayed few histopathological features associated with BRCA1 related tumours.
Conclusions: A comprehensive range of genetic, bioinformatic, and functional analyses have been combined for the characterisation of BRCA1 unclassified sequence variants. Consistent with the functional analyses, the combined odds of causality calculated for the 1706A variant after multifactorial likelihood analysis (1:142) indicates a definitive classification of this variant as “benign”. In contrast, functional assays of the 1708E variant indicate that it is pathogenic, possibly through subcellular mislocalisation. However, the combined odds of 262:1 in favour of causality of this variant does not meet the minimal ratio of 1000:1 for classification as pathogenic, and A1708E remains formally designated as unclassified. Our findings highlight the importance of comprehensive genetic information, together with detailed functional analysis for the definitive categorisation of unclassified sequence variants. This combination of analyses may have direct application to the characterisation of other unclassified variants in BRCA1 and BRCA2.
- BIC, Breast Information Core
- BRCT, BRCA1 C-terminal
- DHPLC, denaturing high performance liquid chromatography
- DMEM, Dulbecco’s modified Eagle’s medium
- ER, estrogen receptor
- FCS, fetal calf serum
- LCL, lymphoblastoid cell line
- LOH, loss of heterozygosity
- LR, likelihood ratio
- PR, progesterone receptor
- SNP, single nucleotide polymorphism
- SNuPE, single nucleotide primer extension
- BRCA1
- functional analysis
- unclassified variants
Statistics from Altmetric.com
- BIC, Breast Information Core
- BRCT, BRCA1 C-terminal
- DHPLC, denaturing high performance liquid chromatography
- DMEM, Dulbecco’s modified Eagle’s medium
- ER, estrogen receptor
- FCS, fetal calf serum
- LCL, lymphoblastoid cell line
- LOH, loss of heterozygosity
- LR, likelihood ratio
- PR, progesterone receptor
- SNP, single nucleotide polymorphism
- SNuPE, single nucleotide primer extension
The pathogenicity of many genetic variants in disease associated genes can be predicted from the nature of the genetic variation. For example, sequence changes that prevent protein expression or that cause loss of important functional domains can be classified as loss of function mutations. However, single exonic nucleotide changes can present a challenge. Such changes have been associated with alterations in transcript stability,1 transcript splicing,2 translation efficiency,1 protein folding,3 protein-protein interactions,4 and the capacity to perform specific cellular functions.4 Interpretation of the pathogenicity of single nucleotide changes is a challenge for the clinical management of many inherited diseases and predispositions including Duchenne muscular dystrophy,5 cystic fibrosis,6 X linked mental retardation,7 and inherited cancer syndromes such as hereditary non-polyposis colon cancer4 and familial breast cancer.8 There is a growing interest in developing efficient and reliable ways to classify the pathogenicity of these variants and a variety of approaches have been reported recently. These include analyses of patterns of co-segregation,9 assessment of variant frequency in unaffected controls,6 predictions based on the position and nature of the amino acid change,7 and biochemical and functional assays.11–28,35
BRCA1 is a breast cancer susceptibility gene encoding a protein of 1863 amino acids with multiple roles in DNA repair, transcriptional activation, apoptosis, and cell cycle regulation (reviewed in Rosen et al29). Pathogenic mutations in the BRCA1 gene have been identified in approximately 15–20% of families with multiple cases of breast and ovarian cancer.30 However, the mechanism(s) by which most mutations in BRCA1 contribute to breast cancer are poorly understood. A further level of complexity is added by the spectrum of unclassified sequence variants described in this gene to date, with over 1000 different unclassified sequence variants in BRCA1 reported on the Breast Information Core (BIC) database alone (http://www.nhgri.nih.gov/Intramural_research/Lab_transfer/Bic/). The pathogenicity of only a small number of these variants has been inferred genetically10,31–34 or tested functionally.11–28,35 Underscoring the interest in and need for accurate classification of sequence variants in BRCA1 are recent descriptions of novel approaches to predict the pathogenicity of non-synonymous amino acid substitutions. These approaches include analysing interspecies sequence variation36,37 and a combination of genetic and bioinformatic predictive investigations.8 This approach offers some advantages over more laborious and expensive experimental analyses, particularly in a clinical setting. How well the data from such predictions correlate with the results of biochemical and functional studies on the same variant, however, is yet to be established.
We have set out to determine the pathogenicity of two sequence variants, G1706A and A1708E, using a number of genetic, bioinformatic, biochemical, and cellular investigations. Although A1708E has been classified as a missense mutation by BIC, the precise molecular defect is not understood, nor has the variant been formally evaluated in the multifactorial model of causality.8 These variants were initially selected for analysis because of their proximity within the C-terminal region of BRCA1, for which several functional assays have been developed. The C-terminal region is a highly conserved structure containing two BRCA1 C-terminal (BRCT) tandem repeat domains. Missense changes within this motif cause protein folding defects18,25 and inhibit transcriptional transactivation.14 The G1706A and A1708E variants are located in the first BRCT domain (amino acids 1650–1736) and have been partially characterised by predictive modelling and some functional assays (reviewed in Mirkovic et al38). Here we present data from a wider variety of approaches that further test these predictions and provide a cellular and molecular basis for pathogenic risk assessment.
METHODS
Genetic analysis
Australian study pedigrees were recruited by the Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab) according to eligibility criteria established by the consortium (http://www.kconfab.org/epidemiology/1eligibility.asp). Two Australian pedigrees in which affected index cases were ascertained to be carriers of the BRCA1 5236G>C (G1706A) variant and one Australian pedigree with the affected index case carrying the 5242C>A (A1708E) variant were selected for analysis. Sequencing or denaturing high performance liquid chromatography (DHPLC) analysis of the coding and flanking intronic regions of BRCA1 and BRCA2 in the index cases revealed no other mutations.
A Spanish pedigree carrying the G1706A variant was ascertained by the Genetics Service, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain. In the index case of this pedigree, the 1706A change was detected by mutation analysis of BRCA1 and BRCA2 using SSCP methodology and sequencing to confirm the nucleotide alteration, and relatives were screened for the variant by sequencing.
Two English A1708E pedigrees were ascertained by the Clinical Genetics Service at Guy’s Hospital in London, United Kingdom. The A1708E variant was identified in the index case of the first pedigree (UK1708E1) by SSCP, followed by sequencing to confirm the mutation. The other member of the family tested was screened for the variant by restriction enzyme analysis. In the second English pedigree (UK1708E2), the A1708E variant was identified in the proband by hydroxylamine fluorescent chemical cleavage of mismatch followed by sequencing confirmation. Subsequently, other carriers within the family were identified by restriction enzyme assay.
In total, 29 individuals from the G1706A pedigrees and 19 individuals from the A1708E pedigrees were genotyped. We estimated the odds for causality associated with these variants using a Bayes factor analysis by maximising the evidence in favour of causality over the hazard ratio, based on the method described by Thompson et al.9 In order to determine the penetrance associated with the variants, we used a modified segregation analysis,39 which estimates cumulative risk to age 70 of breast cancer in carriers (although, due to the small sample size, 95% confidence intervals of cumulative risk may be underestimated), where models were fitted under maximum likelihood theory using the MENDEL statistical package.40 As controls, 180 unaffected females over the age of 45 with no reported family history of breast cancer were recruited through the Australian Breast Cancer Family Study41 and screened by DHPLC for these variants. Variants absent in a sample of 180 individuals are estimated to have an allele frequency with an upper 95% confidence limit less than 1%, the formal definition of a polymorphism. All pedigrees used in the genetic analysis can be supplied by the corresponding author upon request.
Loss of heterozygosity analysis of tumours
Loss of heterozygosity (LOH) at BRCA1 was assessed for index cases by macrodissection of tumour rich (70%) regions of available paraffin sections. DNA was extracted as described previously.42 A PCR product including the 1706 and 1708 positions was generated using the primer pair 5236SeqF (5′-GAGGCTCTTTAGCTTCTTAGGAC-3′) and 5236SeqR (5′-AAACGTTAGGTGTAAAAATGCAA-3′), and sequenced in both directions using the ABI Big Dye terminator system (Applied Biosystems, Foster City, CA, USA) and the forward (5236SeqF) or reverse (5236SeqR) primers. LOH was scored by the significant reduction (<50% peak height relative to normal sequence trace) or absence of the heterozygous peak seen in the germline control.
Single nucleotide primer extension assay
To verify expression of the wildtype and variant BRCA1 transcripts in lymphoblastoid cell lines (LCLs) generated from heterozygous carriers of these variants, total RNA was extracted from LCLs using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and analysed by single nucleotide primer extension (SNuPE).43 A 383 bp fragment of cDNA flanking the 1706 and 1708 positions of BRCA1 was generated by RT-PCR using the Superscript III Reverse Transcriptase kit from Invitrogen and primers Check1F (5′-CCAGAAGAATTTATGCTCGTG) and OR (5′-CAGCTGTACCATCCATTCCA). As positive and negative controls, fragments were also generated with the same primers from the pZeoSV based expression constructs containing wildtype, G1706A, and A1708E BRCA1 cDNA (see below). PCR products were purified using a Qiagen QIAQuick PCR purification kit (Qiagen, Valencia, CA, USA). SNuPE assays were performed on these fragments with radiolabelled dNTPs and the primer pairs SNuPE1706F and SNuPE1706R (5′-GGACACTGAAATATTTTCTAG and 5′-ACCCATTTTCCTCCCGCAATT, respectively) and SNuPE1708F and SNuPE1708R (5′-CTGAAATATTTTCTAGGAATTG and 5′-GCTAACTACCCATTTTCCTCCC, respectively). Radiolabelled fragments were separated on a 10% denaturing acrylamide gel and visualised by autoradiography.
Grantham alignments
Extensive protein multiple sequence alignments were made with the online alignment engines TCoffee and 3Dcoffee.44 The degree of sequence variation present in the alignment was used to calculate the number of positions that are under strong functional constraint or not,37 and the likelihood ratio for whether a substitution at any particular position will be deleterious or not.8 Grantham scores45 were calculated both for each position in the alignments and for each missense mutation versus the canonical human sequence. The relationship between the two Grantham scores was used to determine the fit between the missense substitution and the range of variation observed at its position in the alignment as described previously.8,37 The analysis included 13 full length BRCA1 sequences, the most divergent of which was from the tunicate Ciona.
Protein modelling
Molecular modelling was carried out on the simulated crystal structure of the BRCT repeat region of BRCA1 (JNX1.pdb) that incorporates positions 1706 and 1708,46 using an SGI work station running the Insight II software package (Accelrys, San Diego, CA).
ESE analysis
The wildtype BRCA1 cDNA (NCBI Accession No. U14680) was examined on an exon-by-exon basis for sequences that act as binding sites for the serine/arginine rich family of splicing enhancers using the ESEfinder program (http://rulai.cshl.edu/tools/ESE/). The variant cDNA sequence was then screened by ESEfinder and compared to the wildtype sequence to identify any loss or gain of predicted SR binding sites.
Alternate splicing analysis
Exon 18 sequence containing the 1706A and 1708E variants was analysed for splice acceptor or donor sites using SpliceSiteFinder (http://www.genet.sickkids.on.ca/~ali/splicesitefinder.html). To test for possible alterations in BRCA1 splicing, EBV transformed LCLs generated from variant carriers were treated for 4 h with cycloheximide (100 μg/ml) to stabilise RNA species. RNA was then extracted using the TriPure Isolation Reagent (Roche, Indianapolis, IN, USA). cDNA synthesis was performed and a 345 bp RT-PCR product was amplified using the one step Titan One Tube RT PCR system (Roche) with forward primer 5′ATGCTCGTGTACAAGTTTG 3′ (BRCA1 exon 17) and reverse primer 5′ CTGTGGGCATGTTGGTGAA 3′ (BRCA1 exon 21). Products were visualised on a 2% agarose gel.
Expression plasmids and constructs
A plasmid containing a full length BRCA1 cDNA with three N-terminal c-Myc tags and an in-frame Kozak sequence on a pcDNA3.1 backbone was kindly provided by D Livingston (Dana Farber Cancer Institute, Harvard Medical School, Boston, MA, USA) for subcloning and generation of the mutagenised expression constructs. The BRCA1 cDNA fragment of this parental plasmid was entirely sequenced prior to subcloning, and several single nucleotide polymorphisms (SNPs) (rs1799949, rs799917, rs16940, rs16941, rs16942, and rs4986849) were identified. These SNPs are published on the NCBI and BIC SNP databases and form a common haplotype.47 A fragment containing the c-Myc tags, Kozak sequence, BRCA1 cDNA, and 114 bp of 3′UTR was excised from the parental plasmid with XhoI and ligated into the XhoI site of the pZeoSV vector (Invitrogen). The 1706A and 1708E mutations were generated in this plasmid using the Stratagene QuikChange PCR site directed mutagenesis protocol (Stratagene, La Jolla, CA, USA). Primer pairs for the G1706A mutagenesis protocol were Quik5236F (5′-GACACTGAAATATTTTCTAGCAATTGCGGGAGGAAAATGGG-3′ and Quik5236R (5′-CCCATTTTCCTCCCGCAATTGCTAGAAAATATTTCAGTGTC-3′). The primer pairs for the A1708E mutagenesis protocol were RTW5242F (5′CTGAAATATTTTCTAGGAATTGAGGGAGGAAAATGGGTAGTTAG-3′) and RTW 5242R (5′-CTAACTACCCATTTTCCTCCCTCAATTCCTAGAAAATATTTCAG-3′). The BRCA1 5382insC deletion mutant expression plasmid was kindly donated by Dr B Weber, University of Pennsylvania, USA. Large scale DNA preps were made using the Qiagen Plasmid MaxiKit and the inserts of each plasmid were sequenced entirely to verify their identity.
Trypsin sensitivity assay
A 1571 bp product was amplified by PCR from the pZeo expression constructs containing the wildtype, 1706A, and 1708E variants with a T7 forward primer 5′GGATCCTAATACGACTCACTATAGGAACAGACCACCATGG GTCTGAGTGACAAGGAATT3′ and reverse primer 5′CTGGGGTATCAGGTAGGT3′, encompassing BRCA1 exons 12–24. The products were translated in vitro using the Promega TNT Coupled Reticulocyte Lysates System (Promega, Madison, WI, USA) incorporating 35S methionine (NEN). Protein products were digested for 12 min at 37°C using trypsin (Sigma, St Louis, MO, USA) as described previously25 at concentrations of 0, 0.6, 6, and 60 µg/ml. Protein products were separated by 14% SDS-PAGE and visualised by autoradiography.
Cytoplasmic localisation of ectopic BRCA1 in MCF7 cells
MCF-7 human breast cancer cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), and grown at 37°C in a humidified 5% CO2 atmosphere. Cells were seeded onto sterile glass coverslips and transfected at 50–60% confluency with 1–2 mg of plasmid DNA using Lipofectamine Reagent (Life Technologies, Gaithersburg, MD, USA) according to the manufacturer’s instructions. At 6 h post-transfection, the transfection mix was removed and replaced with DMEM containing 10% FCS. Cells were fixed and processed 30 h post-transfection for fluorescence microscopy. Transfected cells were fixed in 3.7% formalin/PBS for 15 min and processed for immunostaining. Myc tagged ectopic BRCA1 was detected by immunofluorescence using the anti-Myc rabbit polyclonal antibody A-14 (Santa Cruz Biotec, Santa Cruz, CA, USA). Myc antibody was detected with biotin conjugated secondary antibodies (Santa Cruz) and Texas Red-avidin D (Vector Laboratories, Burlingame, CA, USA). Cell nuclei were counterstained with the chromosome dye Hoechst 33285 (Sigma). The subcellular localisation of each ectopic protein was determined by scoring cells using an Olympus BX40 epifluorescence microscope, and the proportion of cells displaying nuclear, nuclear/cytoplasmic, or cytoplasmic staining of BRCA1 was determined as previously described.15 Digital images were collected using a SPOT camera.
Transcriptional transactivation assays
The 1706A and 1708E variants were generated in a pGAL4B expression construct containing the BRCA1 activation domain within a region spanning amino acids 1293–186348 using the Stratagene QuikChange PCR site directed mutagenesis protocol. Primer pairs for generating the 1706A variant were G1706AF 5′-GGACACTGAAATATTTTCTAGCAATTGCGGGAGGAAAATG-3′ and G1706AR 5′-CATTTTCCTCCCGCAATTGCTAGAAAATATTTCAGTGTCC-3′ and primers for the A1708E mutagenesis protocol were RTW5242F/R, described above. Assays were carried out as described previously,48 with two exceptions. MCF7 cells were used instead of 293T cells in the mammalian cell assay and Fugene 6 (Roche) transfection reagent, used according to the manufacturer’s instructions, replaced the calcium phosphate precipitation method of cell transfection.
Western blots
Transiently transfected MCF7 cells were counted and lysate prepared from identical numbers of cells for each transfection. Lysate was electrophoresed, blotted, and probed with FLAG or GAL4 antibodies as described previously.48
Centrosome amplification immunofluorescence and confocal microscopy
Cells were cultured on glass cover slips and transfected with Myc-BRCA1 wildtype and mutant constructs using Fugene 6 according to the manufacturer’s instructions. For indirect immunofluorescence, cells were fixed with cold methanol, permeabilised, and stained with primary anti-pericentrin (1:800) polyclonal (Santa Cruz Biotechnologies) and anti-myc (9E10) (1:200) monoclonal (Santa Cruz Biotechnologies) antibodies 4 days after transfection. Texas Red goat anti-rabbit (1:800) and Oregon Green goat anti-mouse (1:800) secondary antibodies were subsequently added, along with 1 μg/ml Hoechst (Molecular Probes, Eugene, OR, USA). Centrosome numbers were counted in a minimum of 50 Myc expressing cells from each of two independent experiments. For subcellular localisation experiments, 48 h post-transfection the cells were fixed with cold methanol, stained with 1 μg/ml Hoechst (Molecular Probes), anti-myc (9E10) monoclonal (Santa Cruz Biotechnologies) primary antibody, and Oregon Green goat anti-mouse (1:800) secondary antibody. All images were acquired with a Zeiss LSM510 confocal microscope.
Histopathology
Available tumour sections from all the pedigrees were analysed for BRCA1-like histology by pathologists blind to mutation status. Sections were scored for parameters recognised to be associated with BRCA1 tumours.49–54 The BRCA1 mutation associated phenotype was designated as “medullary”, “atypical medullary carcinoma”, or “ductal/no special type”, with high grade, a high mitotic count (>16 mitotic figures/10 high power fields), and one or more of the following features: >25% pushing margin; confluent necrosis; prominent lymphocytic infiltrate. The estrogen receptor (ER) and progesterone receptor (PR) status of the tumour sample was available from some pathology reports.
RESULTS
Genetic and LOH analysis
In Australian G1706A pedigree no. 1 (6-99-006; described in Phelan et al35), two of the three breast cancer patients (ages at diagnosis: 66 and 44 years) and the single ovarian cancer patient (age at diagnosis: 45 years) were found to be 1706A carriers, whereas one woman diagnosed with breast cancer at age 53 was not a carrier.35 The Bayes factor calculated for this family was 0.7086 with a likelihood for causality ratio of 1:1.3. In Australian G1706A pedigree no. 2, the sole surviving breast cancer patient, the single ovarian cancer patient (ages at diagnosis: 49 and 52 years, respectively) and two other unaffected women (aged 70 and 90 years) were found to be 1706A carriers. In this pedigree, the variant was carried by the mother of the index case and not the father whose sister was diagnosed with breast cancer at age 40 years. The Bayes factor calculated for this family was 0.0317, with a likelihood for causality of 1:32. In the Spanish G1706A pedigree, the variant was not carried by the proband’s mother who had a family history of breast and ovarian cancer and therefore was presumed to have been inherited from the father (not tested) who had no known family history of cancer. The Bayes factor analysis for the Spanish G1706A family yielded a value of 0.0196 and a likelihood for causality of 1:51. The likelihood of causality for the 1706A variant using the Bayes factor analysis method for the combined Australian and Spanish families was 1:2274.
In the Australian A1708E pedigree, both living members with breast cancer (ages at diagnosis: 47 and 58 years) were found to carry the 1708E variant. The deceased mother of a carrier was diagnosed with breast cancer at age 47, and a deceased cousin of a carrier was diagnosed with breast cancer at age 30 years. For the Australian 1708E pedigree, the likelihood of causality was 2.8:1.
In the English pedigree UK1708E1, the variant was identified in the proband (breast cancer diagnosed at age 28) and in the mother of the proband (ovarian cancer diagnosed at age 55). No other individuals in this family were genotyped. The Bayes factor analysis for this pedigree yielded odds of causality of 2.0:1. In the pedigree UK1708E2, two A1708E variant carriers were identified. One carrier was diagnosed with breast cancer at age 39 years. The other carrier has not been diagnosed with cancer to date. Two other individuals from this pedigree without breast cancer were tested and found to be negative for the A1708E variant. The Bayes factor analysis for this pedigree yielded a ratio of 0.7982:1, equating to an odds of causality of 1:1.3. The total Bayes factor for the combined Australian and English A1708E pedigrees was 4.5:1 in favour of causality, and the estimated penetrance for A1708E was 100% (95% CI 98% to 100%) to age 70 years.
Neither the 1706A nor 1708E variants were detected in any of the 180 control samples. Loss of the wildtype allele was demonstrated in tumour tissue from one case from the Australian G1706A no. 1/6-99-006 and A1708E pedigrees (data not shown). However, in the tumour sample isolated from the Spanish G1706A pedigree, no LOH was detected. No LOH data were available from the United Kingdom A1708E pedigrees.
SNuPE assay
The relative expression of wildtype and variant alleles was measured using the SNuPE assay. Wildtype and variant alleles were expressed at approximately equal levels (fig 1) indicating that the variant sequences do not affect the stability of their respective transcripts.

RNA stability of BRCA1 variants G1706A and A1708E in carriers from Australian pedigrees. SNuPE assays were carried out on homozygous wildtype (wt) and variant control plasmid PCR products, and RT-PCR products from RNA isolated from LCLs generated from BRCA1 variant carriers. (A) G1706A SNuPE assays in forward and reverse directions show approximately equivalent stability of transcripts containing the wt (G) and variant (C) nucleotides at nt5236. (B) A1708E SNuPE assays in forward and reverse directions show approximately equivalent stability of transcripts expressing the wildtype (C) and variant (A) nucleotides at nt5242 of the BRCA1 mRNA.
ESE and splice variant analysis
The ESEfinder program was used to predict the effects of the 1706A and 1708E variants on serine-arginine rich splicing factor binding to consensus exonic enhancer sequences. While this program may not identify all ESEs, and splicing predictions may not be definitive,28 replacement of the wildtype BRCA1 sequence with the 1706A variant in exon 18 was predicted by ESEfinder to disrupt a consensus binding site for the SC35 splicing enhancer. ESEfinder modelling of the 1708E variant predicted disruption of SC35 and SRp55 splicing enhancer binding sites. Further, SpliceSiteFinder analysis of the 1708E variant predicted the creation of an additional acceptor sequence. However, RT-PCR analysis revealed no evidence of aberrant splicing (data not shown). Thus, the 1706A and 1708E variants did not appear to affect the splicing of their respective primary transcripts, supporting previous findings for 1706A by Campos et al.28
In silico analysis and protein modelling
Both variants occur in a highly conserved region of the BRCT domain of BRCA1. Analysis of the degree of cross-species variation at the positions of these two substitutions predicted that both are likely to be deleterious. Position 1708 is invariant, resulting in a likelihood ratio (LR) of 58:1 in favour of causality for 1708E. The Grantham score for the substitution alanine>glutamic acid is 107. This score is above the average Grantham score for all possible missense single nucleotide substitutions in BRCA1 (n = 78), tending to reinforce the expectation that the substitution is deleterious. Exactly one substitution is observed at position 1706, from which we calculate an LR of 16:1 in favour of causality for 1706A. Interestingly, the cross-species substitution at 1706 is glycine>alanine in the tunicate Ciona (SVT, unpublished data). This means that the human substitution 1706A is outside the range of variation observed in vertebrates but within the range of variation observed from human to tunicate. There are no firm data from which to determine whether, or by how much, that observation modifies the likelihood that 1706A is deleterious. But we also note that the Grantham score for glycine>alanine is 60, which is below the average Grantham score for all possible missense single nucleotide substitutions in BRCA1. Coupled with the observation of alanine at the corresponding position in the tunicate sequence, consideration of the Grantham scores would tend to decrease the likelihood that the 1706A substitution is actually high risk.
Protein structure modelling predicted that the substitution of an alanine at position 1706 would cause a moderate increase in hydrophobicity and size. The glycine at position 1706 occurs within an α-helix structure running from leucine 1701 to isoleucine 1707. Substitution of glycine with a larger alanine at 1706 was predicted to disrupt a bend in the helix, possibly with functional consequences if normal protein function is reliant on the maintenance of this conformation. The substitution was predicted to cause clashes between the side chains of valine 1687 and valine 1713, which were not relieved by changes in the side chain rotamers of each residue.
The 1708E variant was predicted to cause significant disruption of protein structure, with a shift from a highly hydrophobic residue (alanine) to a larger and strongly hydrophilic residue (glutamic acid). Examination of the BRCA1 crystal structure showed that the alanine, glycine, and glycine residues at positions 1708, 1709, and 1710, respectively, form a tight turn structure likely to permit only small residues as replacements. Substitution of the alanine with the much larger glutamic acid residue at position 1708 was predicted to be incompatible with maintaining the turn structure. Furthermore, this substitution was predicted to produce clashes, mostly between the carboxyl group of glutamine at 1708 and the methionine at 1783 in the second BRCT domain. Clashes predicted for the glutamate residue were all with the main chain (peptide) atoms and the γ-methylene group of the methionine, where the clashes involved distances that were too small and unfavourable interactions between hydrophobic and charged hydrophilic atoms. Changes in the side chain rotamers of each residue were not predicted to relieve the clash.
Proteolytic degradation assays
Consistent with the conformational changes predicted by protein modelling for 1708E, resistance of this variant protein fragment to trypsin degradation was reduced compared to wildtype and 1706A proteins. Degradation of the 1708E protein fragment by trypsin occurred at a 10-fold lower concentration of the enzyme than required for the 1706A and wildtype BRCA1 fragments (fig 2), and supports previous findings showing that the 1708E variant was less resistant to proteolytic digestion.25 The wildtype and 1706A variant BRCA1 proteins showed similar degradation profiles, indicating that the minor structural change predicted for the 1706A variant protein did not have a measurable effect on the stability of the BRCA1 protein in these assays.

The BRCA1 variant 1708E is significantly more susceptible to proteolytic digestion than the wildtype or 1706A isoforms of BRCA1. BRCA1 cDNA fragments (wildtype, 1706A, 1708E) were translated in vitro and digested with increasing concentrations of trypsin.
Subcellular localisation of ectopically expressed variant and wildtype BRCA1
The 1706A and 1708E sequence variants were introduced into myc tagged full length BRCA1 cDNA expression constructs, which were then used to transfect various cell lines for expression and functional analysis. In MCF7 cells transiently transfected with these constructs, normal nuclear localisation of the transiently expressed wildtype and 1706A proteins was indicated by ubiquitous staining in both the nuclear and cytoplasmic compartments after staining with myc antibodies for ectopic BRCA1 (fig 3). In contrast, cytoplasmic mislocalisation of the BRCA1 1708E protein was observed. This predominantly cytoplasmic expression of the 1708E variant is similar in pattern to that previously described for another cancer associated BRCA1 variant, 5382insC.15 However, while the nuclear localisation of the 5382insC variant was shown to be increased to wildtype levels with the co-transfection of BARD1,15 in these experiments BARD1 expression did not restore nuclear localisation of the 1708E variant to a wildtype pattern of expression. Nuclear localisation of the wildtype and 1706A constructs was significantly enhanced by the co-transfection of BARDI.

The BRCA1 1708E variant is mislocalised in MCF7 cells. MCF7 cells were transiently co-transfected with myc tagged BRCA1 cDNA (wt, wildtype; 1706v, 1706A; 1708v, 1708E) and YFP or YFP-BARD1 expression constructs. Ectopic BRCA1 expression was analysed by c-Myc immunofluorescence.
Transcriptional transactivation assays
As the 1706A and 1708E variants map to the region of BRCA1 implicated in transcriptional transactivation, the potential effect of these sequence changes on this function of BRCA1 was investigated by introducing these changes into a cDNA construct encoding amino acids 1293 to 1863. In transiently transfected MCF7 cells, regulation of transcriptional transactivation by the 1706A variant resulted in reporter activity indistinguishable from wildtype levels (fig 4A). In contrast, reporter construct activity in cells transfected with the cDNA construct containing 1708E was similar to that of the empty vector, suggesting minimal capacity for this variant protein to transactivate transcription. Western blot analysis of total protein indicated that levels of the 1708E protein were significantly lower than the wildtype and 1706A proteins (fig 4B) suggesting that the reduced transcriptional transactivation was likely to reflect aberrant expression rather than function of the BRCA1 protein.

Transcriptional transactivation of reporter plasmids in vitro by 1708E BRCA1 is defective in a mammalian system. (A) Mammalian MCF7 cells co-transfected with BRCA1 expression constructs show no significant difference in reporter activity between the cells transfected with the wtBRCA1 or 1706A variant BRCA1, but a significant decrease with the 1708E variant. Values are the mean and standard deviations of three independent experiments. (B) Western blot expression analysis of MCF7 cells transiently transfected with BRCA1 constructs and probed with FLAG antibody. Arrow denotes BRCA1(wt or mutants)-Gal4DBD fusion protein. Protein levels are normalised for even loading. Markers shown are in kDa.
Centrosome amplification
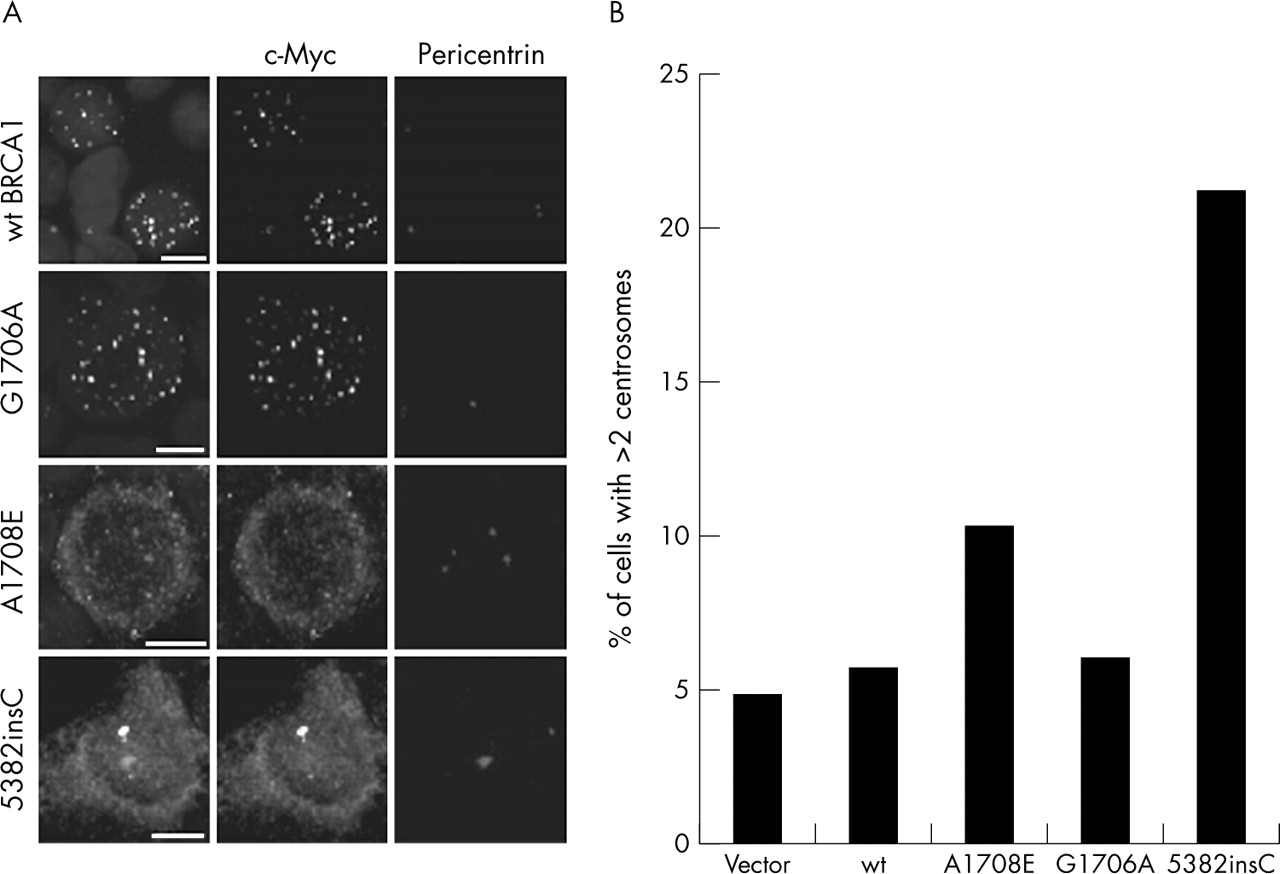
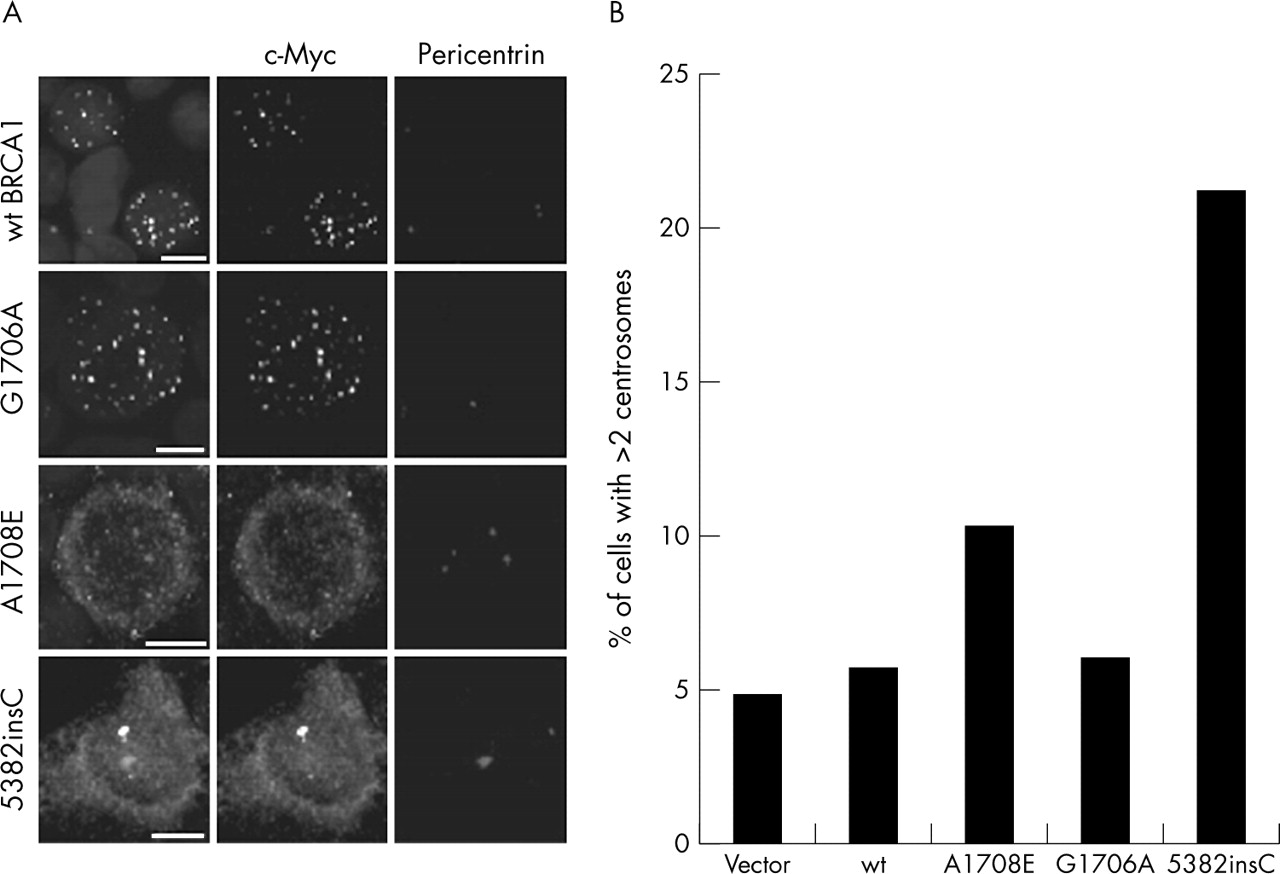
Another important nuclear function of BRCA1 is regulation of centrosomes (reviewed in Starita et al55). To address the consequences of the two variants on this function of BRCA1, 293T cells were transfected with wildtype and variant constructs, followed by centrosome analysis and counting. Centrosome amplification was evident in cells transfected with the BRCA1 1708E variant (fig 5) but not in those transfected with the 1706A and wt BRCA1 constructs. Staining for c-Myc in the 293T cells also indicated that the 1708E variant is largely mislocalised to the cytoplasm and not transported to the nucleus, supporting data presented earlier (fig 3) for MCF7 cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Increased centrosome amplification is observed in 293T cells transiently transfected with the BRCA1 1708E expression plasmid. (A) 293T cells transfected with BRCA1 expression constructs containing the 1706A and 1708E variants and the 5382insC deletion mutant were stained with c-Myc to identify cellular localisation of the ectopic constructs and pericentrin to identify centrioles. Scale bar represents 10 nm. Results show exclusion of 1708E from the nucleus. (B) The number of 293T cells containing more than two centrosomes indicating centrosome amplification was counted for each of the transient transfections of the various expression plasmids. Negative control is vector and wildtype BRCA1. Positive control is BRCA1 5382insC. Results show a significant increase in the number of cells showing centrosome amplification in the 1708E transfection.
Tumour pathology
The pathology of tumours from the 1706A and 1708E carriers in the Australian pedigrees was strikingly different (table 1). The two tumours examined from 1708E carriers had many BRCA1 related features. In contrast, the histopathology of the 1706A tumour from the Australian pedigree no. 1/6-99-006 index case did not have a BRCA1 related tumour histological profile. Similarly, the Spanish G1706A pedigree tumour sample did not display features consistent with a BRCA1 defect (table 1).
Histopathology of BRCA1 1706A and 1708E breast tumours
DISCUSSION
Using genetic and bioinformatics analyses, functional assays, and histopathology, we have attempted to determine the pathogenicity of the BRCA1 1706A and 1708E variants, and to assess these approaches for their potential use in the characterisation of other BRCA1 and BRCA2 unclassified variants.
Several functional assays indicated a severe functional abrogation of BRCA1 proteins carrying the 1708E variant. Protein modelling of the 1708E variant predicted a significant folding defect. Proteolytic digestion assays by us and others25 supported this prediction by showing that a protein fragment containing the 1708E variant was significantly more susceptible to degradation in vitro. Pathogenicity of the 1708E variant was further supported by assays showing cytoplasmic mislocalisation of ectopically expressed BRCA1 protein carrying the 1708E variant in MCF7 and 293T cells. Cytoplasmic mislocalisation has been demonstrated in MCF7 cells for several other BRCA1 BRCT domain mutants of known pathogenicity, including the 5382insC, M1775R, and Y1853X mutations.15
The failure of co-transfected BARD1 to restore nuclear import of 1708E BRCA1 in MCF7 cells suggests that BARD1 is unable to complex with mutant BRCA1. The mechanism by which BARD1 and BRCA1 work together is currently being elucidated15,56–58 and although these two proteins interact via the N-terminal RING domain of BRCA1, it is possible that variant sequences in the C terminus of BRCA1 affect the three dimensional protein structure. The predicted 1708E conformational change may induce an altered tertiary structure masking the BARD1 binding region and preventing the formation of the BRCA1/BARD1 heterodimer. This would impact on both the nuclear import and ubiquitin ligase functions of BRCA1. Alternatively, it is possible that the conformational change is sufficiently severe in terms of charge and size that nuclear import is physically impeded.
Cytoplasmic mislocalisation of the 1708E variant BRCA1 may impede its function as a regulator of transcription. Indeed, transcriptional transactivation by the 1708E variant was significantly lower than by the wildtype protein in our assays. These data are consistent with previously reported disruption of BRCA1 transcriptional transactivation by the 1708E variant in mammalian59 and yeast based assays.60
In the centrosome amplification assays, the 1708E variant displayed features consistent with pathogenicity. Centrosome amplification has been associated with over 70% of invasive breast tumours61 and has been shown to be a major cause of mitotic defects in cancer cells leading to chromosomal instability, cell cycle checkpoint dysregulation, and loss of tissue differentiation (reviewed in D’Assoro et al62). The C terminus of BRCA1 has been implicated as an important mediator of centrosome regulation, through the formation of a ubiquitin ligase complex with BARD1 for the ubiquitination of γ-tubulin, a major centrosome component involved in the maintenance of centrosome number. Defects in the ubiquitination of γ-tubulin induced by overexpression of the N-terminal γ-tubulin binding fragment of BRCA1 in COS 7 cells63 or by expression of mutant γ-tubulin in mammary Hs578T cells55 caused centrosome amplification. C-terminal deletion mutants of BRCA1 also disrupted γ-tubulin ubiquitination.55 Our experiments showed that, in the MCF7 breast cell line, the 1708E variant was not localised to the nucleus with endogenous or ectopic BARD1, indicating that formation of the BRCA1/BARD1 complex was significantly diminished. We suggest that the centrosome defect associated with the 1708E variant may in part be a consequence of dysregulated ubiquitination of γ-tubulin and possibly other centrosome associated proteins.
In support of the functional data, the histopathology review of the two 1708E carrying tumours identified several features commonly associated with BRCA1 mutation carrying tumours. Evolutionary sequence alignment analysis gave odds in favour of causality of 58:1 for 1708E. However, the genetic evidence of pathogenicity from the A1708E pedigrees is weak, giving an odds of causality of 4.5:1. Applying the multifactorial likelihood model8 to assess pathogenicity, the combined odds from these two approaches was 262:1, which does not meet the criteria of Goldgar et al8 of 1000:1 for a pathogenic variant. Nevertheless, the odds are consistent with the functional assays which indicated that this variant has features of a high risk mutation.
In contrast, the protein stability, transcriptional transactivation, and subcellular localisation of the 1706A variant were all comparable to the wildtype in our assays. Protein modelling of the 1706A variant predicted only a mild conformational change, and this prediction was borne out experimentally, with a proteolytic digestion profile similar to the wildtype protein. This variant also showed minimal effects on subcellular localisation, centrosome amplification, and transcriptional transactivation. Our transcriptional assay data contrasts with evidence from another research group,35 who reported that cells expressing 1706A showed a moderately reduced transcriptional transactivation capacity compared to wildtype BRCA1 in both mammalian and yeast systems. However, these inconsistencies may be due to differences in cell lines and the length of BRCA1 cDNA used.
The histopathology review of the G1706A carrying tumour from the Australian and Spanish pedigrees did not have a BRCA1 associated tumour phenotype. The loss of the wildtype allele in this tumour may simply reflect background LOH, which is estimated to be about 30% in breast tumours.64,65 Sequence alignment analysis gave odds in favour of causality of 16:1 for 1706A. The Bayesian odds of causality for this variant in the combined Australian and Spanish pedigrees studied was 1:2274. Using data from sequence alignment and co-segregation analysis, the combined odds of causality after applying the multifactorial likelihood model is 1:142. This meets the criteria of Goldgar et al8 of 1:100 for a neutral variant, consistent with the functional assays indicating that this variant is not a high risk mutation.
In conclusion, we have presented genetic, histopathological, and functional evidence to suggest that the A1708E variant of BRCA1 is pathogenic, as classified by BIC, possibly through subcellular mislocalisation. However, this variant remains formally designated as unclassified according to the criteria of Goldgar et al8 after application of the multifactorial likelihood model using sequence alignment and co-segregation data. In contrast, analysis of the G1706A variant in functional assays suggests that it is unlikely to be associated with significant risk of breast cancer and may be classified as benign according to the criteria of Goldgar et al.8 While classification of 1708E and other variants may ultimately be achieved by analysis of further co-segregation data, and inclusion of LOH and control frequency data in the multifactorial likelihood model, we suggest the application of an appropriate selection of the functional assays described here and elsewhere will enhance predictions of pathogenicity of unclassified sequence variants in BRCA1 and BRCA2. Assay selection will largely be determined by the location of unclassified sequence variants within known structural or functional motifs in the gene. In our hands, the cytoplasmic mislocalisation, proteolytic sensitivity, and centrosome amplification assays were found to be the most informative for the functional analysis of the G1706A and A1708E variants in the BRCT domain of BRCA1. These assays may also be useful for variants affecting other regions of the protein. In light of the diversity of roles played by BRCA1 at the cellular level, it is unlikely that any single assay will conclusively characterise sequence variants in this gene. Where possible, a comprehensive approach utilising genetic, bioinformatic, and functional analyses to classify sequence variants is most likely to reliably indicate potential pathogenicity, and may eventually also assist in delineating variants with moderate risk from those with high or low/no risk.
Acknowledgments
The authors would like to thank the kConFab research nurses and staff for data collection, Heather Thorne, Lynda Williams, and Dani Surace for DNA preparation, Jan Groves for establishment of the LCLs, Eveline Niedermayr for supplying data, Don Roberts and Anna Marsh for technical assistance, the staff of the Familial Cancer Clinics for their support of kConFab, and the families for their participation. We also thank ABCFS researchers Margaret McCredie and Graham Giles for their roles in the establishment of the ABCFS, Gillian Dite for database management, and Sarah Steinborner and Deon Venter for preparation of DNA.
REFERENCES
Footnotes
-
Published Online First 2 June 2005
-
kConFab has been funded by the Kathleen Cuningham Foundation, National Breast Cancer Foundation, National Health and Medical Research Council (NHMRC), Cancer Council of Victoria, Cancer Council of South Australia, Queensland Cancer Fund, Cancer Council of New South Wales, Cancer Foundation of Western Australia, and Cancer Council of Tasmania. This research was supported by a grant from the Susan G. Komen Breast Cancer Foundation, the NHMRC, and the INHERIT BRCAs programme from the Canadian Institute for Health Research. ABS is funded by an NHMRC Career Development Award. JH, GC-T, and BRH are NHMRC Senior Principal, Principal, and Senior Research Fellows, and WA is funded by an NHMRC Dora Lush postgraduate scholarship. Collection of ABCFS control DNA was funded by the NHMRC
-
Competing interests: none declared