Article Text

Abstract
Oral–facial–digital syndrome type 1 (OFD1) is characterised by an X linked dominant mode of inheritance with lethality in males. Clinical features include facial dysmorphism with oral, tooth, and distal abnormalities, polycystic kidney disease, and central nervous system malformations. Large interfamilial and intrafamilial clinical variability has been widely reported, and 18 distinct mutations have been previously reported within OFD1. A French and Belgian collaborative study collected 25 cases from 16 families. OFD1 was analysed using direct sequencing and phenotype–genotype correlation was performed using χ2 test. X inactivation studies were performed on blood lymphocytes. In 11 families, 11 novel mutations, including nine frameshift, one nonsense, and one missense mutation were identified, which spanned nine different exons. A combination of our results with previously reported cases showed that the majority of mutations (65.5%) was located in exons 3, 8, 9, 13, and 16. There was phenotype–genotype correlation between (a) polycystic kidney disease and splice mutations; (b) mental retardation and mutations located in exons 3, 8, 9, 13, and 16; and (c) tooth abnormalities and mutations located in coiled coil domains. Comparing the phenotype of the families with a pathogenic mutation to families with absence of OFD1 mutation, polycystic kidneys and short stature were significantly more frequent in the group with no OFD1 mutation, whereas lingual hamartomas were significantly more frequent in the group with OFD1 mutation. Finally, an X inactivation study showed non-random X inactivation in a third of the samples. Differential X inactivation between mothers and daughters in two families with high intrafamilial variability was of particular interest. Slight phenotype–genotype correlations were established, and X inactivation study showed that skewed X inactivation could be partially involved in the pathogenesis of intrafamilial clinical variability.
- CNS, central nervous system
- OFD1, oral–facial–digital syndrome type 1
- PKD, polycystic kidney disease
- OFD1
- dominant X linked mode of inheritance
Statistics from Altmetric.com
Oral–facial–digital syndrome type 1 (OFD1; OMIM #311200) or Papillon–Léage–Psaume syndrome belongs to the heterogeneous group of OFD syndromes. Incidence is 1/50 000 live births. Clinical features include hypertelorism, broad nasal bridge, hypoplasia of the nasal alae, small median cleft or pseudocleft of the upper lip (45%), hyperplastic buccal frenulae, cleft palate (80%), bifid or lobulated tongue (30–45%), lingual hamartomas (70%), hypodontia, especially frequent on low lateral incisors, and facial asymmetry. Abnormalities of the fingers (45%) are more frequent than toes (25%), and include brachydactyly, syndactyly, clinodactyly, and more rarely, polydactyly.1,2 Polycystic kidney disease is common (50%).3,4 Central nervous system malformations are variable (40%), including corpus callosum agenesis, arachnoid cysts, cerebellar abnormalities, hydrocephalus, and porencephaly, and may be associated with mental retardation (40%).5,6 Characteristic radiological features are represented by irregular pattern of radiolucency and/or spicule-like formation in metacarpals and phalanges on hand x rays. Clinical variability is high and diagnosis may be difficult in minor cases. In addition, the phenotype overlaps with those reported in the other 11 forms of OFD syndromes.7,8,9,10 OFD1 is distinguished by its X linked dominant mode of inheritance with lethality in males and common association with polycystic kidney disease.3,4,11,12 The OFD1 gene, also named Cxof5 (Xp22.2–22.3), contains 23 coding exons.13–16OFD1 is ubiquitously expressed in adult tissues, and in vitro studies have shown that it seems to escape X inactivation.16 Eighteen mutations have been reported in the literature.15,17–19 This French and Belgian collaborative study collected 25 observations of patients with OFD1 from 16 families. Clinical, molecular, and genotype–phenotype correlation studies are presented.
PATIENTS AND METHODS
Patients
Patients’ clinical data and samples were collected from genetic clinics through a French and Belgian collaboration. Female patients with oral, facial, and digital features, associated or not with a family history compatible with X linked dominant mode of inheritance, were included. Patients with an OFD syndrome other than type 1, or with familial history ruling out dominant X linked pattern, were excluded. Family 1 was referred for prenatal diagnosis of porencephaly, and clinical details have been reported in detail previously.20 The other 15 families were ascertained through the association of facial dysmorphism, oral features, and/or cerebral, renal, and digital malformations.
Methods
Mutation analysis
Blood samples and informed consents were collected from 24/25 patients. DNA was extracted from peripheral EDTA, according to previously reported methods.21 In the female fetus reported in family 1, DNA was also extracted from fetal blood, frozen liver, lung, fibroblasts, thymus, and paraffin block for the tongue. Mutations in OFD1 were screened by using primer sequences covering all 23 exons.15 PCR amplifications of the samples were run through 30 cycles consisting of 40 seconds at 94°C (denaturation), 40 seconds at 50°C or 55°C (annealing), and 40 seconds at 72°C (extension), with a final extension step of 10 minutes. Sequencing of PCR products was performed using the ABI Prism7 BigDye Terminator Cycle Sequence kit (version 1.1; Applied Biosystems, Foster City, CA, USA) in both ranges, and analysed using an ABI Prism7 3100 Genetic Analyzer, according to the manufacturer’s instructions. Identification of a mutation in a proband was followed by direct sequencing of the affected exon in each family member. When a missense mutation was found, 100 French control DNAs were sequenced for the region of interest in order to exclude a polymorphism.
Linkage studies
In familial cases in which no pathogenic mutations were found, DNA was amplified by PCR using fluorescent primers flanking microsatellite polymorphisms of the region of interest. The primers were chosen according to their location on the Marshfield map, as previously used for linkage studies.22 Primer sequences were obtained from the National Center for Biotechnology Information database. The selected markers used were DXS1224 and DXS7108, located telomeric to OFD1 (0.59 and 16.56 cM respectively) and DXS7105 and DXS8018, located centromeric to OFD1 (11.19 and 12.64 cM respectively). Microsatellite analysis was undertaken using the ABI 3700 Genescan/Genotyper system, according to the manufacturer’s instructions. Genotypes were recorded manually.
Genotype–phenotype correlation
The mutations found in the present study were added to those previously described in the literature. A correlation between some clinical features (hypertelorism, cleft lip and palate, buccal frenulae, lingual hamartomas, tooth abnormalities, brachydactyly, syndactyly, polydactyly, polycystic kidney disease, corpus callosum agenesis, mental retardation), mutation types (frameshift, splice mutations), and mutation locations (coiled coil domains, exons 3, 8, 9, 13, and 16) was studied using a χ2 test. The study was not performed when the sample was insufficient to be contributive for genotype–phenotype correlation (n<5). The correlation for the same clinical features between the group of patients with a pathogenic OFD1 mutation and patients with absence of OFD1 mutation was also studied using a χ2 test.
X chromosome inactivation
The X chromosome inactivation pattern in lymphocytes was studied in 24 female cases. The FRAXA locus and the human androgen receptor assay were used with experimental conditions modified from the literature.23–26 Primer sequences and experimental conditions are available on request.
RESULTS
Clinical studies
From 16 families, 25 affected female cases were collected, average age 22 years. Eight cases were sporadic. Pedigrees of the familial cases are presented in fig 1. Clinical features of the whole series are summarised in tables 1–3. Facial dysmorphism was a consistent feature, including buccal frenulae (76%), hypertelorism (67%), hypoplasia of the nasal alae (58%), cleft palate or lip (56%), lingual hamartomas (52%), lobulated or cleft tongue (48%), downslanting palpebral fissures (50%), and pseudocleft of the upper lip (21%). Available clinical pictures are presented in fig 2. Tooth abnormalities (44%) included hypodontia and malposition. Distal abnormalities with predominant brachydactyly (64%) were more frequent in hands (64%) than in feet (28%) (fig 3). Associated malformations included mainly corpus callosum agenesis (71%) and polycystic kidney (44%). Mild to moderate mental retardation was present in 11/23 cases (48%), but information was not available in 2/25 cases (fetus and 3 week old newborn). Additional cerebral features, including arachnoid cysts (4/14), cerebellar hypoplasia (3/14), hydrocephalus (2/14), and porencephaly (1/14) were more rarely associated with OFD1 syndrome. Short stature was also described (4/25). Hand x rays were performed in only four cases, and showed spicule-like formations in metacarpals and/or phalanges in three of those.
Clinical features of the reported patients with OFD1 mutation
Clinical features of the reported patients without OFD1 mutation
Clinical features of the reported patients with and without OFD1 mutation

Pedigrees of the familial cases reported in this article.
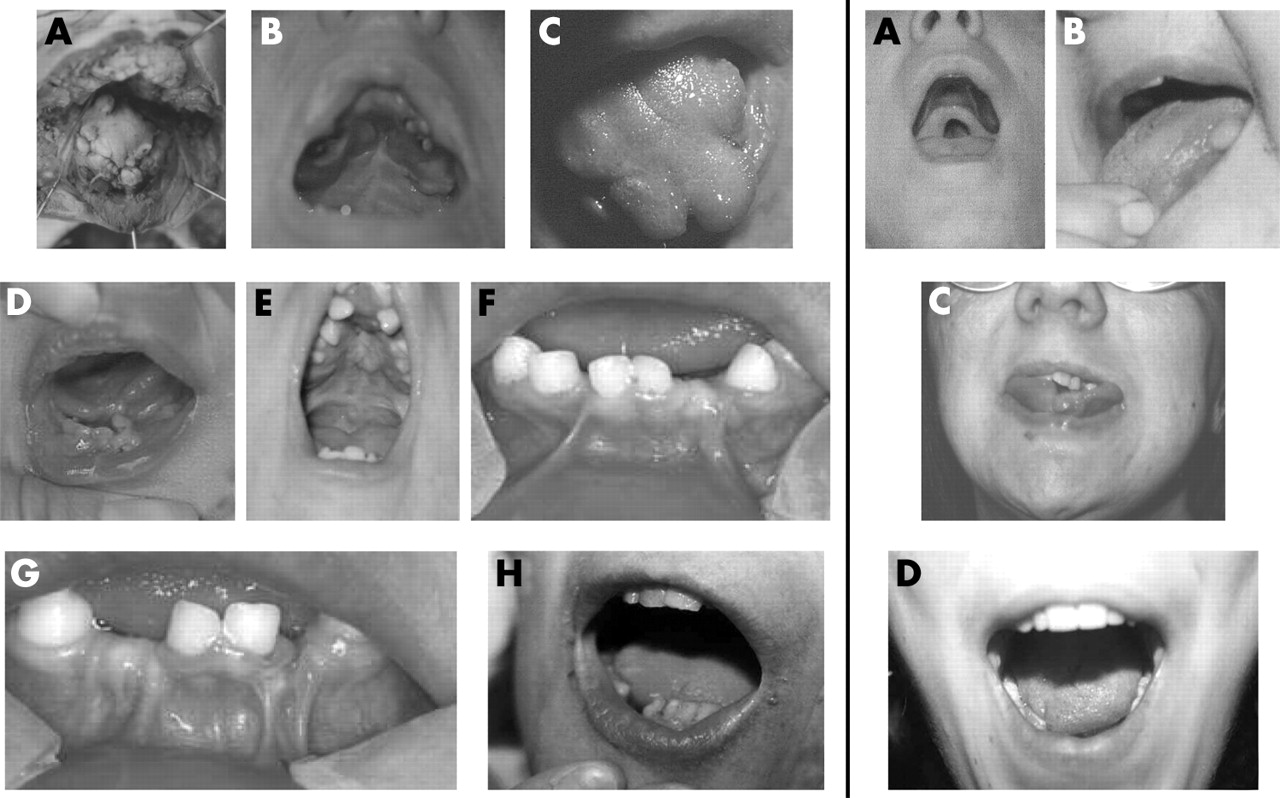
Clinical pictures of the reported patients showing oral abnormalities. Left side, patients with mutations identified in OFD1: (A) Individual IV-3, family 1 with buccal frenulae and lingual hamartoma. (B, C) Proband of family 4 with cleft and lobulated tongue, buccal frenulae, and surgery for cleft palate. (D) Proband of family 5 with cleft and lobulated tongue, buccal frenulae, and lingual hamartoma. (E) Proband of family 6 with cleft and lobulated tongue, buccal frenulae, lingual hamartoma, narrow palate, tooth abnormalities, and surgery for cleft palate. (F, G) Twin 1 and 2, family 8 with buccal frenulae, hypodontia, and abnormally shaped teeth. H) Proband of family 9 with lobulated tongue, hypodontia, and abnormally shaped teeth of the inferior maxilla. Right side, patients without mutations in OFD1: (A) Individual IV-2, family 12 with cleft palate. (B) Individual III-2, family 12 with lingual hamartoma. (C) Individual III-3, family 13 with cleft and lobulated tongue and dental misposition. (D) Proband of family 16 with cleft and lobulated tongue. Permission from the patients was obtained for publication of these photographs.

{kind=link}
{kind=link}
{kind=link}
Clinical pictures of the reported patients showing distal abnormalities. Left side, patients with mutations identified in OFD1: (A) Individual IV-3 in family 1 with postaxial polydactyly. (B) Individual III-3 in family 1 with brachydactyly predominant on the fourth and clinodactyly on the third finger. (C) Proband of family 4 with severe brachydactyly predominant on the fourth finger and clinodactyly. (D, E) Proband of family 5 at 7 months and at 11½ years of age, with brachydactyly on fingers III-IV and clinodactyly. (F) Proband of family 6 with severe generalised brachydactyly predominant on finger I and clinodactyly. (G) Proband of family 11 with brachydactyly. Right side, patients without mutations in OFD1: (A) Individual III-2, family 12 with brachydactyly, clinodactyly, and low set and deviated left thumb. (B) Individual III-3, family 13 with brachydactyly, clinodactyly, and syndactyly. (C) Individual II-2, family 13 with severe brachydactyly. (D) Proband of family 14 with severe brachydactyly predominant on finger IV. (E) Proband of family 16 with brachydactyly and syndactyly. Permission from the patients was obtained for publication of these photographs.
Mutation analysis
Eleven novel mutations in OFD1 were found in eight sporadic and three familial cases. Mutations located in nine different exons included eight deletions and one insertion (nine frameshift), one stop mutation, and one point mutation (table 4). In family 6, a G138S missense mutation was found, and was therefore tested in 100 controls. In families 12 and 15, the same base exchange was identified 295 bases proximal to the start codon, and was also found in an unaffected relative male, indicating a polymorphism. No mutation was found in five patients, including three familial cases (families 12, 13, and 15) and two sporadic cases (cases 14 and 16).
OFD1 mutations identified in our series
Linkage studies
In family 12, microsatellite polymorphism analysis of the Xp22.2-22.3 region showed compatible linkage with the OFD1 region, as all affected individuals had inherited the same chromosome X.
Genotype–phenotype correlation
Renal cysts were significantly more frequently associated with splice mutations compared with other mutations (χ2 = 0.012). Mental retardation was significantly more frequently associated with mutations located in exons 3, 8, 9, 13, and 16 compared with mutations located in other exons (χ2 = 0.04). Tooth abnormalities were significantly more frequently associated with mutations located in coiled coil domains compared with mutations with other locations (χ2 = 0.007). No correlation was found between OFD1 clinical features and frameshift compared with other mutations.
When comparing the phenotype of the 11 families with a pathogenic mutation with that of the five families with absence of mutation within OFD1 (table 3), we showed that there was no significant difference between the presence or absence of hypertelorism, cleft lip and palate, buccal frenulae, tooth abnormalities, digital abnormalities, corpus callosum agenesis, or mental retardation in both groups. However, short stature was only seen in the group of patients with no OFD1 mutation, and polycystic kidney disease was significantly more frequent in the group with absence of OFD1 mutation compared with the group with a pathogenic mutation (χ2 = 0.035). Conversely, lingual hamartomas were less common in the group with absence of mutation compared with the group with a pathogenic OFD1 mutation (χ2 = 0.041).
X chromosome inactivation
X chromosome inactivation results in lymphocytes could not be produced in 1/24 patients because of homozygosity at both loci (patient II-2, family 1). Analyses showed non-random inactivation in 7/23 patients (30%) from six families (table 5), three of which were familial cases. Determination of the parental origin of the skewed allele was possible in two families. In family 1, the skewed X chromosome was of maternal origin in patient III-3 (mother) and of paternal origin in patient IV-3 (fetus). X chromosome inactivation studies in fetal tissues were not successful because of poor quality of the DNA. In family 15, the skewed X chromosome was of paternal origin in patient III-2.
X chromosome inactivation studies in our series
DISCUSSION
We report a large French and Belgian clinical and molecular study of 25 patients from 16 families presenting with suspected OFD1 syndrome. Mutations in OFD1 were identified in 16/25 patients.
The frequencies of the major clinical features were similar to those reported in the literature. Facial dysmorphism was a consistent feature, and included hypertelorism (67%), buccal frenulae (76%), hypoplasia of the nasal alae (58%), lingual hamartomas (52%), and cleft palate or/and lip (56%). Tooth abnormalities (44%), including hypodontia and malposition were not always seen in association with cleft palate and lip; 2/11 patients without cleft palate or/and lip presented noticeable tooth abnormalities. Considering distal abnormalities, brachydactyly involving predominantly the second, third, and fourth fingers was characteristic (64%).
Recently, the increasing use of renal ultrasound scan revealed that polycystic kidney disease (PKD) is commonly associated with OFD1 syndrome.3,4 Our study confirms these data, as PKD was present in 7/16 cases (44%). However, 8/9 patients were too young (from fetal period to 11.5 years of age) to draw definite conclusions about the absence of kidney involvement.
The central nervous system (CNS) may also be involved in as many as 40% of the cases.5 In our group, corpus callosum agenesis (10/14 cases) was more frequent than previously reported. However, results may be biased, as cerebral magnetic resonance imaging was not systematically performed, particularly when mental retardation was absent. Moderate to mild mental retardation was frequent (48%), but could not always be correlated with CNS malformations. Spicule-like formations in metacarpals and/or phalanges were present in 3/4 cases. This previously reported sign is frequent and specific to OFD1 syndrome but hand x rays were rarely performed (4/25 cases).
The majority of clinical features overlaps with those reported in the other OFD syndromes.27 Clinical classification remains imperfect. For example, OFD7 syndrome has been previously reported in only one family with renal involvement and mother to daughter transmission.28 Later, the birth of a new affected female baby with corpus callosum agenesis in the third generation suggested OFD1 syndrome. Subsequent linkage studies at the OFD1 locus showed compatible linkage despite a non-significant lod score (1.7).22 The authors suggested that OFD7 should not be a distinct clinicopathological entity. OFD8 syndrome (OMIM #311200), characterised by polydactyly and tibial dysplasia, is described as a recessive X linked heritable disease, but this type could also be allelic with OFD1.29
Direct sequencing of OFD1 in 16 families revealed 11 different mutations. In 5/16 families, no mutation was found. The detection level of 67% in our series is higher than previously reported (50%).22 Surprisingly, the detection rate was higher in sporadic (80%) than in familial cases (50%), although compatible with dominant X linked inheritance (families 13 and 15), with the presence of spicule-like formation metacarpals and/or phalanges in family 15. Similarly, linkage was compatible with the OFD1 locus in family 12, associated with PKD and corpus callosum agenesis. In the two sporadic cases without identified mutation (cases 14 and 16), the presence of PKD strongly suggests OFD1 syndrome. In such cases, complementary molecular studies would be necessary to detect intronic mutations, mutations within the promoter, or large rearrangements such as deletions, as recently described in a recent report.30 When comparing the phenotype of the families with or without a pathogenic OFD1 mutation, we found that the presence of polycystic kidneys and short stature and the absence of lingual hamartomas suggested the absence of an OFD1 mutation, although such data could not be useful at an individual level.
There have been 18 OFD1 mutations reported in the literature; we identified a further 29 in this study, including 15 deletions, four insertions, two nonsense, five missense, and three splice mutations.15,17–19 No mutation was found in exons 18–23. The majority of mutations occurred in exons 3, 8, 9, 13, and 16 (19/29 cases, 66%), suggesting that these exons may represent regions for mutational hotspots. This hypothesis has previously been postulated for exons 3, 13, and 16.18
Mutational analysis of OFD1 may be of interest for diagnosis and genetic counselling, especially in sporadic cases. Indeed, in family 2, family history is both compatible with dominant X linked and autosomal recessive mode of inheritance (OFD2 syndrome) with pseudodominance because of familial consanguinity. Identification of an out of frame deletion in exon 16 confirms OFD1 syndrome and therefore a risk of transmission to daughters of 50%. Another example is given by family 5. The mother presented with isolated bifid uvula, and identification of a nonsense mutation (L144X) only in the proband confirmed a de novo mutation and a consequent low risk of recurrence in other children, limited to the risk of gonadal mosaicism.31
When analysing the phenotype–genotype correlations, mental retardation and cleft lip/palate were reported more frequently in association with mutations in exons 3, 8, 9, 13, and 16 compared with mutations in other exons. PKD was more frequently associated with splice mutations compared with other mutations, and tooth abnormalities more frequently associated with mutations in coiled coil domains compared with other domains. Unfortunately, no correlation between CNS abnormalities and mutations within the LisH motif could be tested because of the small size of the sample.
Skewed X inactivation was found in 7/23 cases (30%) from six families, a higher rate than in the general population (10%).32 Skewed X inactivation was found in four cases from three families: in two severe cases, the paternal allele was inactivated, and in a moderate case, the maternal allele was inactivated. In family 2, the mother presented a moderate phenotype with skewed inactivation of unknown origin (parental origin could not be identified because of de novo mutation and unavailability of parental DNA samples), whereas her daughter has a severe phenotype with random inactivation. These results suggest that non-random X inactivation may in part explain intrafamilial clinical variability in these three families. In the three sporadic cases with non-random X inactivation, mental retardation and PKD were present in 2/3 cases, also suggesting that non-random X inactivation could explain in part the severe phenotype in sporadic cases. Additional studies in other tissues and in other patients would be of interest.
In conclusion, we report here the identification of 11 new mutations in OFD1 in 16 French and Belgian families who presented with OFD1 syndrome. Slight phenotype–genotype correlations were identified, and the X inactivation study showed that skewed X inactivation could be partially involved in the pathogenesis of intrafamilial clinical variability. These results should be confirmed on larger samples.
ELECTRONIC DATABASE INFORMATION
Online Mendelian Inheritance in Man (OMIM), http://www.nih.nlm.nih.gov/Omim/. National Center for Biotechnology Information datadase, http://www.ncbi.nlm.nih.gov/. Marshfield map, http://www.marshmed.org/genetics/.
Acknowledgments
This work was supported by a grant from the University Hospital of Dijon (Appel d’Offre Interne 2002) and by the Conseil Régional de Bourgogne. We thank the Centre d’Investigation Clinique et d’Epidémiologie Clinque (CIC-EC) for its help in the statistical analysis and J P Ramot for photographic assistance.
REFERENCES
Footnotes
-
Competing interests: none declared