Article Text

Abstract
Background: The major factors individually reported to be associated with an increased frequency of CDKN2A mutations are increased number of patients with melanoma in a family, early age at melanoma diagnosis, and family members with multiple primary melanomas (MPM) or pancreatic cancer.
Methods: These four features were examined in 385 families with ⩾3 patients with melanoma pooled by 17 GenoMEL groups, and these attributes were compared across continents.
Results: Overall, 39% of families had CDKN2A mutations ranging from 20% (32/162) in Australia to 45% (29/65) in North America to 57% (89/157) in Europe. All four features in each group, except pancreatic cancer in Australia (p = 0.38), individually showed significant associations with CDKN2A mutations, but the effects varied widely across continents. Multivariate examination also showed different predictors of mutation risk across continents. In Australian families, ⩾2 patients with MPM, median age at melanoma diagnosis ⩽40 years and ⩾6 patients with melanoma in a family jointly predicted the mutation risk. In European families, all four factors concurrently predicted the risk, but with less stringent criteria than in Australia. In North American families, only ⩾1 patient with MPM and age at diagnosis ⩽40 years simultaneously predicted the mutation risk.
Conclusions: The variation in CDKN2A mutations for the four features across continents is consistent with the lower melanoma incidence rates in Europe and higher rates of sporadic melanoma in Australia. The lack of a pancreatic cancer–CDKN2A mutation relationship in Australia probably reflects the divergent spectrum of mutations in families from Australia versus those from North America and Europe. GenoMEL is exploring candidate host, genetic and/or environmental risk factors to better understand the variation observed.
- ARF, alternate reading frame
- CMM, cutaneous malignant melanoma
- MPM, multiple primary melanoma
- melanoma
- CDKN2A
- multiple primary melanomas
- pancreatic cancer
Statistics from Altmetric.com
The CDKN2A (MIM# 600160) gene is the major known high-risk cutaneous malignant melanoma (CMM) susceptibility gene. CDKN2A encodes two distinct proteins translated, in alternate reading frames (ARFs), from alternatively spliced transcripts. The α transcript encodes the p16 protein; the smaller β transcript specifies the alternative product p14ARF. Germline CDKN2A mutations have been observed in approximately 20–40% of melanoma-prone families from around the world.1 Several variables have individually been reported to be associated with an increased frequency of CDKN2A mutations, including increased number of patients with melanoma, early median age at melanoma diagnosis, the occurrence of pancreatic cancer in a family and the occurrence of multiple melanoma tumours in a patient.2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17 These four features have been assessed separately. However, it has not been previously possible to compare the four factors across geographical regions, nor to examine them simultaneously. Such an evaluation would facilitate interpretation of risks by clinicians from different continents and give a view on putative relationships between genotype and latitude of residence.
The Melanoma Genetics Consortium GenoMEL (http://www.genomel.org), comprising major familial melanoma research groups from North America, Europe, Australia and the Middle East, conducted a study to explore the relationship between these four attributes and the presence of a CDKN2A mutation. All GenoMEL member groups with eligible families participated in this study. The resultant sample of 385 families, 150 of which had CDKN2A mutations, allowed simultaneous evaluation of the four features, as well as inspection of differences by continent.
METHODS
Patients and design
Seventeen GenoMEL centres participated in this study. Families with at least three confirmed patients with melanoma who were screened for mutations in CDKN2A (exons 1α, 2 and 3) were eligible for the study. Mutation evaluation, predominantly sequencing or denaturing high performance liquid chromatography followed by sequencing, was conducted at each centre. All families (n = 385) with and without identified mutations were included. CDKN2A mutations that altered the p16 protein were included in the evaluation; a subset of these mutations also influenced p14ARF.
Table 1 presents the number of families and total number of patients with melanoma by study centre. For all centres, written informed consent was obtained from the subjects before participation in the study under Institutional Review Board-approved protocols. The precise methods of ascertainment for the eligible families differed between groups. Details of the participating families from each centre are described elsewhere (see table 1 for references). All melanoma diagnoses were confirmed by review of histological materials, pathology reports, medical records or death certificates. Only patients with confirmed melanomas (invasive or in situ) were included. This restriction may have resulted in differences for some centres in reported numbers of families presented previously. For each family, the absence/presence and type of CDKN2A mutation were reported. Other variables included number of patients with CMM in each family, age at first melanoma diagnosis for each patient, whether or not a patient with CMM had one versus multiple melanoma tumours, and the number of patients with melanoma or first-degree relatives of patients with melanoma in each family who had pancreatic cancer (81% confirmed).
Number of families, patients with cutaneous malignant melanoma and families with CDKN2A mutations by study centre and summarised by country/continent and overall (total)
Statistical analysis
Using the data provided, we derived four factors for analysis: number of patients with CMM in a family (#CMM/family), number of patients with CMM in a family with multiple primary melanoma (MPM) tumours, median age at CMM diagnosis in a family (MedAge), and number of patients with pancreatic cancer in a family, combining the reports of pancreatic cancer in patients with melanoma and in first-degree relatives of patients with melanoma. These four factors were evaluated across all 17 GenoMEL groups (total) and within specific geographical regions defined by continent (Australia, Europe and North America). In addition, the four attributes were also individually assessed in the European groups, for which there were sufficient numbers of families (ie, Sweden (Lund+Stockholm), Mediterranean Europe (Barcelona+Genoa+Emilia-Romagna), UK (Leeds+Glasgow), The Netherlands (Leiden) and France (Paris)). The non-parametric Wilcoxon–Mann–Whitney test, Fisher’s exact, Jonckheere–Terpstra or Kruskal–Wallis tests, as implemented in the computer programme StatXact (StatXact-4, V.4.0.1), were used to evaluate each of the four factors individually. Multivariate evaluation of the four factors in all families (total) and in families from Australia, Europe or North America was conducted using unconditional logistic regression as implemented in the program Stata (Stata 8.2, V.8.2). It was not possible to evaluate the four factors jointly in groups smaller than that in a continent. For the logistic regression analysis, the presence/absence of a CDKN2A mutation was the dependent variable. We thus measured the association between the “risk” of a CDKN2A mutation and the four factors using backward and forward stepwise logistic regression analyses. In addition, the final models were evaluated using likelihood ratio tests. All statistical tests were two sided.
RESULTS
Table 1 presents the number of families, number of patients with melanoma and number of families with CDKN2A mutations by participating centre. There were 385 families with 1720 patients with CMM in this study. Overall, 39% of families (n = 150) had mutations. The frequency of mutations ranged from 20% (32/162) in Australia to 45% (29/65) in North America to 57% (89/157) in Europe. Table 2 shows the CDKN2A mutations by continent and whether the mutation altered p14ARF.
Number of families with each mutation in CDKN2A by continent
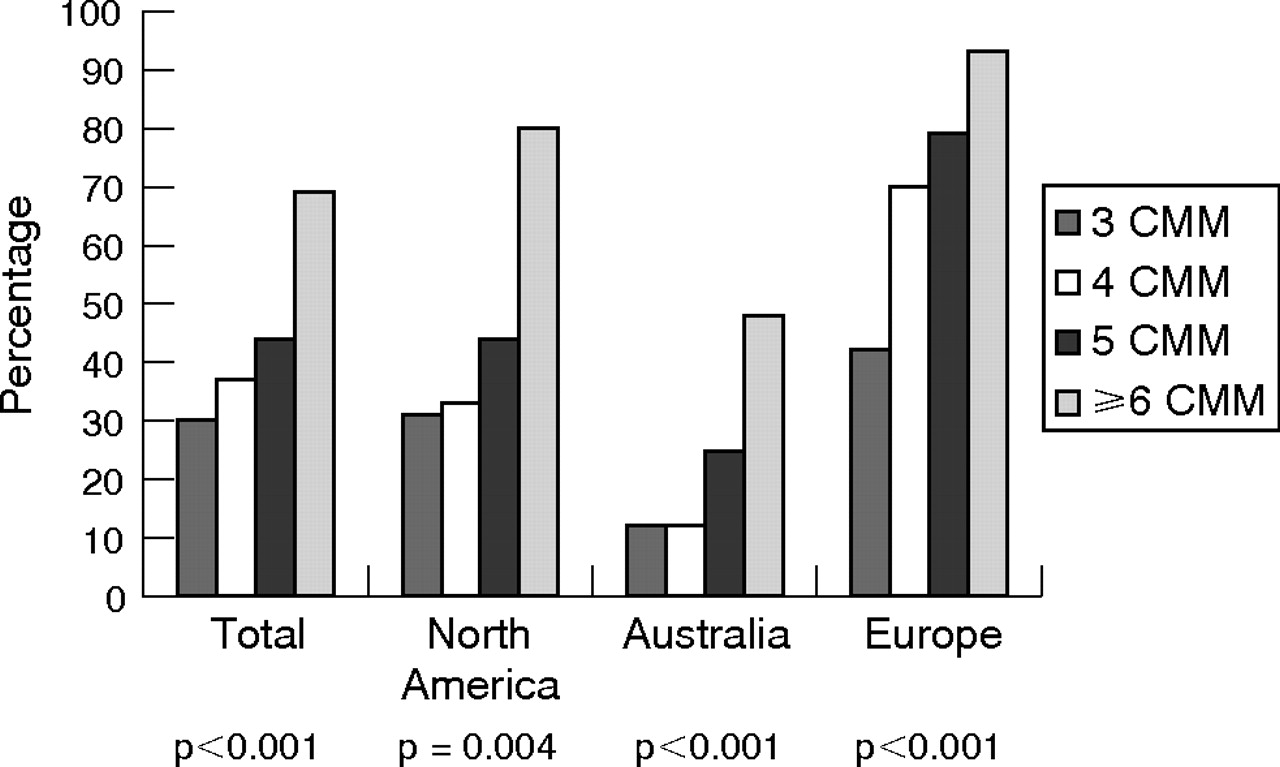
Figures 1–4 show the percentage of families with CDKN2A mutations by number of patients with CMM/family (fig 1), MPM (fig 2), pancreatic cancer (fig 3) and MedAge (fig 4) for each of the four groups (total, North America, Australia and Europe). The frequency of mutations increased significantly as the number of CMM/family increased in each of the four comparison groups (fig 1, p values ranging from p<0.001 to p = 0.004). In addition, among the mutation-positive families, there were significant differences in the distribution of the number of patients with melanoma per family across continents (p = 0.002). In North American and Australian families, the largest increase in mutation frequency was from those families with 5 patients with CMM/family to those with ⩾6 CMM/family (from 4/9 (44%) to 12/15 (80%) and from 6/24 (25%) to 12/25 patients with (48%), respectively). By contrast, in Europe, the greatest increase in mutation frequency was among families with ⩾4 patients with CMM/family (77%) relative to those with 3 patients with CMM/family (42%). Although all European groups showed patterns consistent with the overall Europe result, only families from Sweden showed a significant association between a mutation and number of patients with melanoma per family (p = 0.001).

Percentage of families with CDKN2A mutations by number of patients with cutaneous malignant melanoma per family (no. of CMM/family: 3, 4, 5 and ⩾6) for each of the four groups (total, North America, Australia and Europe). p Values are shown for each of the four groups.
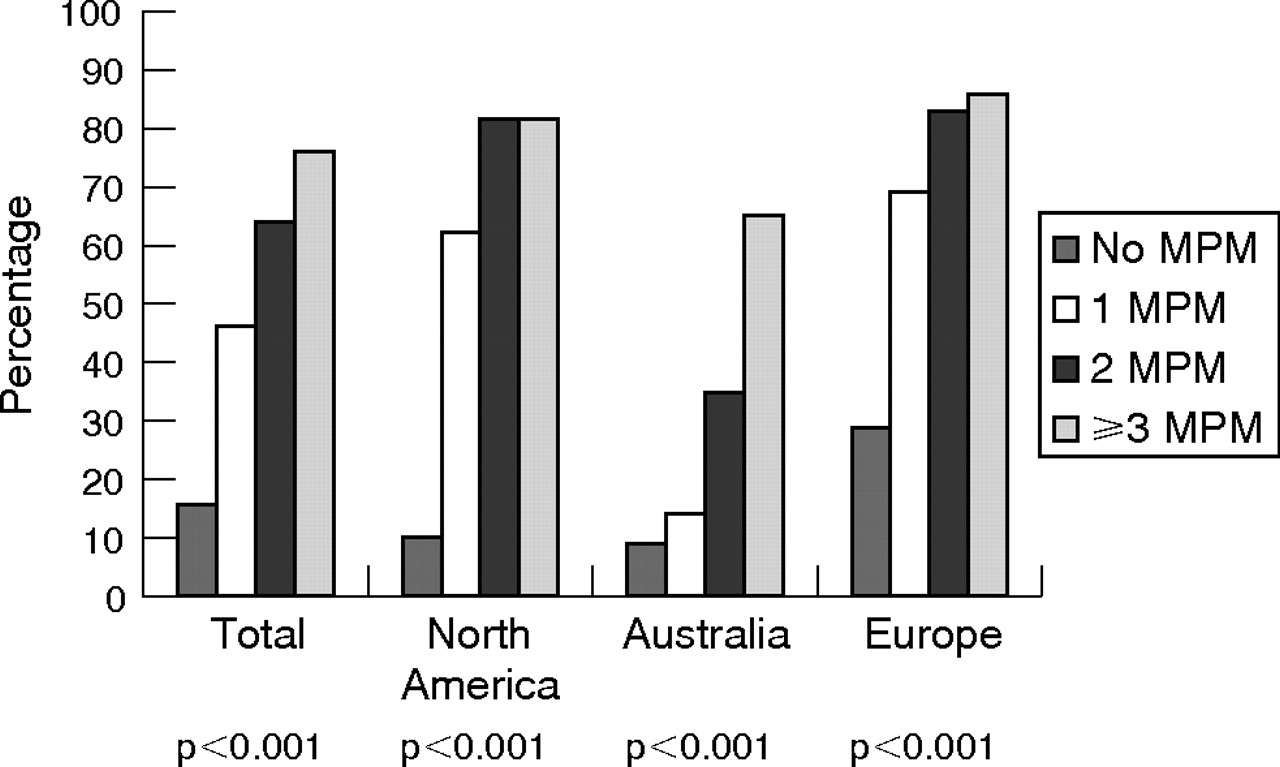
Percentage of families with CDKN2A mutations by number of patients with multiple primary melanoma (MPM: 0, 1, 2 and ⩾3) in a family for each of the four groups (total, North America, Australia and Europe). p Values are shown for each of the four groups.

Percentage of families with CDKN2A mutations by number of patients with pancreatic cancer (0, 1 and ⩾2) in a family for each of the four groups (total, North America, Australia and Europe). p Values are shown for each of the four groups.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Percentage of families with CDKN2A mutations by median age (in years) at melanoma diagnosis (MedAge: <34, 34–40, >40–50 and >50 years) in a family for each of the four groups (total, North America, Australia and Europe). p Values are shown for each of the four groups.
The frequency of mutations increased significantly as the number of patients with MPM in a family increased for all groups (fig 2, p<0.001). We also found significant differences in MPM distribution across continents in mutation-positive families (p = 0.012). The presence of one patient with MPM produced a dramatic increase in mutation frequency for North American (0 patients with MPM: 3/30, 10% v ⩾1 patient with MPM: 26/35, 74%) and European families (0 patients with MPM: 18/63, 29% v ⩾1 patient with MPM: 71/94, 76%) The MPM patterns observed in individual European groups were all similar, but significant associations between MPM and mutations were observed only for families from Sweden (p<0.001), France (p = 0.003) and Mediterranean Europe (p = 0.006). By contrast, for Australian families, a striking increase in the mutation frequency required ⩾2 patients with MPM (⩽1 patient with MPM: 13/122, 11% v ⩾2 patients with MPM: 19/40, 48%).
The relationship between pancreatic cancer and CDKN2A mutations showed differences across groups (fig 3). Overall (total), 72% of families with one reported patient with pancreatic cancer had mutations (31/43) and 81% of families with ⩾2 patients with pancreatic cancer had mutations (13/16). There was a significant association between pancreatic cancer and mutations in families from the total (p<0.001), North America (p = 0.02) and Europe (p<0.001). However, pancreatic cancer was not associated with mutations in Australia (p = 0.38). Only three of nine Australian families (33%) with pancreatic cancer had mutations. By contrast, for North American and European families, ⩾75% of families with at least one patient with pancreatic cancer had mutations (9/12 and 32/38 families, respectively). In the European families, the strongest evidence for an association between pancreatic cancer and CDKN2A mutations came from The Netherlands (p = 0.006), France (p = 0.007) and Sweden (p = 0.04).
Figure 4 shows the relationship between median age at melanoma diagnosis in a family (MedAge) and CDKN2A mutations using quartiles (<34, 34–40, >40–50 and >50 years) defined among the total. All groups showed a significant decrease in the frequency of mutations as MedAge increased. Again, in the families positive for CDKN2A mutation, there was a significant difference in the distribution of MedAge across continents (p = 0.002). The significance resulted from an older median age at melanoma diagnosis in European mutation-positive families compared with either Australian (p = 0.002) or North American (p = 0.03) families. For European families, a mutation frequency >60% was observed when MedAge was ⩽50 years. For Australian families, however, a high mutation frequency (>35%) required an earlier median age at melanoma diagnosis (ie, ⩽40 years). North American families showed a pattern intermediate between Europe and Australia, with a step-function decrease in mutation frequency as MedAge increased.
We compared the distributions of #CMM/family, MPM, pancreatic cancer and MedAge across the most frequent CDKN2A mutations, defined as mutations that occurred in ⩾5 families (c.225_243del19, p.M53I, p.G101W, p.R112-L113insR, c.IVS2-105A>G, p.R24P, c.-34G>T, c.32_33ins9-32 and p.V126D). At least 70% of families with each of these mutations had ⩾1 patient with MPM. For seven of the nine mutations, most of the families had early MedAge. Only p.R112–L113insR and c.-34G>T had <50% of families with MedAge ⩽40 years. The percentage of families with ⩾5 patients with CMM/family ranged from 20% to 80%, with five of nine frequent mutations having <40% of families with ⩾5 patients with CMM/family. However, there were no significant differences in #CMM/family (p = 0.14), MPM (p = 0.40) or MedAge (p = 0.14) across the nine most frequent mutations. By contrast, the distribution of pancreatic cancer differed significantly across these mutations (p<0.001). No families with p.M53I (0/14), c.IVS2-105A>G (0/10) or c.32_33ins9–32 (0/6) mutations had pancreatic cancer; 25–36% of families with p.R24P (2/8), c.-34G>T (2/7) or p.G101W (5/14) mutations had pancreatic cancer; finally, pancreatic cancer was observed in ⩾60% of families with p.R112–L113insR (7/11), c.225_243del19 (12/20) or p.V126D (3/5) mutations. The distribution of pancreatic cancer in families with the most frequent CDKN2A mutations suggested an association with p14ARF (41% of families with a mutation that altered p14ARF had pancreatic cancer v 19% of families with a mutation that did not affect p14ARF). However, the observed pattern was not completely consistent. p.M53I, a mutation that alters p14ARF had no pancreatic cancer in 14 families; similarly, 60% of families with p.V126D had pancreatic cancer, yet this mutation does not alter p14ARF. Additional studies are needed to further evaluate this relationship.
Table 3 shows the multivariate predictors of a CDKN2A mutation for all families (total) and by continent. Given the significant differences observed between continents, we did not constrain the exposed and unexposed/referent categories in the multivariate analysis of the four features to be identical in the continent-specific analyses. There were substantial differences observed across continents. In all groups (total), MedAge⩽50 years, ⩾1 patient with MPM and ⩾1 patient with pancreatic cancer in a family were significant joint predictors of a CDKN2A mutation. However, #CMM/family was not an independent predictor for mutation risk. For the Australian families, ⩾6 patients with melanoma in a family, ⩾2 patients with MPM and MedAge⩽40 years simultaneously predicted the risk of a mutation. As was observed in the univariate analyses, pancreatic cancer in a family did not influence the risk for a CDKN2A mutation. For North American families with the smallest sample size of 65 families, only MedAge⩽40 years and ⩾1 patient with MPM were concurrent predictors of a CDKN2A mutation. Finally, for European families, all four factors jointly predicted the mutation risk (table 3). However, the levels for MedAge (⩽50 years) and #CMM/family (⩾4) were less stringent than that seen in the other comparison groups.
Predictors of a CDKN2A mutation from simultaneous evaluation of the four factors (MedAge, multiple primary melanomas, pancreatic cancer, no of cutaneous malignant melanomas/family) across the groups (Total, Australia, North America and Europe)
DISCUSSION
GenoMEL explored the relationship between select risk factors and presence of a CDKN2A mutation in 385 families with at least three confirmed patients with CMM. Individual examination of each of the four attributes showed that for all comparison groups examined (total, North America, Australia and Europe), there were significant associations between CDKN2A mutations and number of patients with melanoma in a family, the occurrence of multiple primary melanoma tumours and median age at melanoma diagnosis in a family. For pancreatic cancer, all groups except Australia showed significant associations with mutations. The frequencies of mutations and the effects of the four attributes, however, varied significantly across continents. Similarly, joint evaluation of the four features showed different predictors of mutation risk across the geographical areas.
Melanoma incidence rates vary widely among Caucasian populations around the world. The incidence rates are highest in Australia (38.5/100 000 for men; 29.5/100 000 for women), intermediate in North America (16.4/100 000 for men; 11.7/100 000 for women) and generally lowest in Western Europe (7.3/100 000 for men; 10/100 000 for women; CANCERMondial: http://www-dep.iarc.fr).45 Differences in the amount of exposure to ultraviolet radiation, the predominant environmental risk factor for melanoma and variation in host characteristics (eg, hair colour, eye colour, melanocytic nevi, freckling and skin type) may contribute to the wide geographical variation in melanoma incidence rates. In this study, the lowest frequency of CDKN2A mutations was observed in Australia (20%), the area with the highest incidence rates. By contrast, the highest frequency of mutations was observed in Europe (57%), the region with the lowest incidence rates. The underlying difference in incidence rates for melanoma between Australia and Europe is further reflected by the categorisation of the four features in the multivariate analyses. In Australian families (n = 162) in which higher rates of sporadic melanoma occur and fair skin and intense sun exposure predominate, ⩾2 patients with MPM, MedAge⩽40 years and ⩾6 patients with melanoma in a family were needed to concurrently predict CDKN2A mutation risk. For the 157 European families, the factor levels were less stringent (ie, broader), consistent with the lower melanoma incidence rates. Specifically, for the European families, only one patient with MPM, only ⩾4 patients with melanoma in a family and a median age at melanoma diagnosis up to 50 years, in addition to ⩾1 patient with pancreatic cancer, jointly predicted the risk of a CDKN2A mutation. The variation in frequency of CDKN2A mutations by number of patients with melanoma in a family, presence of patients with multiple primary melanoma tumours or pancreatic cancer, and median age at melanoma diagnosis across geographical regions may reflect different distributions of host, genetic or environmental risk factors.
In the univariate analyses, pancreatic cancer was significantly associated with CDKN2A mutations in all regions studied except Australia. As smoking is a well-known risk factor for pancreatic cancer,46 reported differences in smoking patterns with lower rates in Australia compared with most of Western Europe47 may contribute to some of the differences seen. However, the rates of smoking in Sweden are lower than that observed in Australia, and pancreatic cancer is strongly associated with CDKN2A mutations in families from Sweden.12 In addition, there is little difference in pancreatic cancer incidence rates across the regions involved in the current study. The age-standardised incidence rates across the regions range from 6–8/100 000 for men to 5–7/100 000 for women.45 Alternatively, different distributions of mutations observed in Australia versus other areas may influence the association with pancreatic cancer. Most of the common mutations observed in families from Australia (p.M53I, c.IVS2–105A>G, p.R24P or p.L32P) have generally shown low frequencies of pancreatic cancer even in non-Australian populations. In the current study, no Australian families with p.M53I (n = 5), c.IVS2–105A>G (n = 3) or p.R24P (n = 3) mutations had pancreatic cancer. Conversely, two of three families with p.L32P had pancreatic cancer. Further evaluation of the relationship between these common mutations and pancreatic cancer showed that no non-Australian families with p.M53I (0/9) or c.IVS2–105A>G (0/7) had pancreatic cancer. Yet, 40% (2/5) of families with p.R24P from the UK (2/3) and France (0/2) and 1/1 family from the UK with p.L32P had pancreatic cancer. In addition, the CDKN2A mutations (p.R112–L113insR, c.225_243del19, p.V126D and p.G101W) showing the strongest associations with pancreatic cancer in this and previous studies (summarised in Goldstein5) were essentially absent from Australia. Only one Australian family in the current study had any of these four mutations. Thus, the lack of a pancreatic cancer–CDKN2A mutation relationship in Australian families may reflect the spectrum of mutations observed in Australia versus Europe and North America. This finding needs to be confirmed.
The current study had several limitations. The ascertainment and sampling of families at most of the GenoMEL centres was not population-based, and each centre obtained data on extended family members to different degrees. Additionally, the study sample was restricted to families with at least three patients with melanoma, thereby selecting for families at higher risk of melanoma compared with either population-based cases or unselected case series.17,48 The divergent ascertainment across study sites could, in consequence, have produced variation in the distribution of the four factors of interest. Nonetheless, in the families without CDKN2A mutations, there were no significant differences in the distribution of median age at melanoma diagnosis (p = 0.54), patients with multiple primary melanoma tumours (p = 0.39) or pancreatic cancer (p = 0.24) across the families from North America, Europe or Australia. However, the number of patients with melanoma in a family varied, with European families having significantly fewer patients with melanoma than Australian (p<0.001) and North American (p = 0.009) families. This difference probably reflects the lower incidence rates of melanoma in Western Europe, and consequently fewer families with large numbers of patients with melanoma. Moreover, it was not possible to fully evaluate the four attributes in areas smaller than a continent. Although continent was selected as the geographical region under study, there is variation in melanoma incidence rates, particularly in the European centres that were part of this study. As such, a continent may not adequately separate families with different risks of melanoma and hence, distributions of risk factors. Finally, as the study was restricted to melanoma-prone families with at least three confirmed patients with CMM, the results from the study may not be applicable to families with only one or two patients with melanoma.
Additional studies are needed to expand the findings from the current study to a wider spectrum of patients with melanoma by evaluating simultaneously the four features examined here in patients with melanoma unselected for a positive family history. A clinic-based study from Boston and Toronto16 assessed the predictors of a CDKN2A mutation in unselected patients with melanoma, and showed that number of patients with melanoma in a family, median age at melanoma diagnosis and multiple melanoma tumours in a patient were the most important predictors for a mutation. These findings are generally consistent with those observed in the current study. However, given the different joint predictors of mutation risk observed in families from each of the three continents, larger sample sizes and greater geographical diversity are needed to extend these findings to multiple geographical regions. Furthermore, comparison of results from multiple-case families (eg, the current study) and geographically comparable population-based or unselected series of patients could extend the interpretation of risks to different geographical regions and advance the understanding of the relationships between genotype and latitude of residence.
By working together, GenoMEL has produced a dataset to allow simultaneous evaluation of four factors previously individually associated with an increased frequency of CDKN2A mutations. Identification of such features enhances our ability to understand the differential risks that contribute to melanoma. The analyses showed differences in the frequency of CDKN2A mutations according to the number of patients with melanoma in a family, the occurrence of multiple CMM tumours in a patient, early median age at melanoma diagnosis in a family, and the occurrence of pancreatic cancer in a family across geographical regions. The differences may reflect distinct host, genetic or environmental risk factors. GenoMEL is exploring candidate risk factors to better understand the variation observed.
Acknowledgments
We thank the participating families, whose generosity and cooperation have made this study possible. We also thank the nurses, doctors, scientists and other health professionals who referred or evaluated patients with melanoma and families for this study.
REFERENCES
Footnotes
-
Published Online First 11 August 2006
-
Funding: This research was supported in part by the intramural research programme of the National Institutes of Health, NCI, DCEG and NHGRI. This work was also partially supported by research grants 1 RO1 CA83115-01A2 “Genetic epidemiology of melanoma” from the National Cancer Institute (DEE); RO1 CA88363 from NCI (NH), National Health and Medical Research Council of Australia (Brisbane group); R01 CA102422 from NCI (LACA), the Tom C Mathews Jr Familial Melanoma Research Clinic, the Huntsman Cancer Foundation (Utah group); Cancer Research UK (Leeds group); grants 01/1546 and 03/0019 from Fondo de Investigaciones Sanitarias, V2003-REDC03/03 and /07, a personal grant to Francisco Cuellar CONACYT, Personal Scholarship 152256/158706, Mexico City, Mexico (Barcelona group); FIRB RBNE0149752001 and COFIN 2004/061840_003 (GB-S; Genoa group); The Swedish Cancer Society: research grants 4860-B05-03XAC, 5012-B05-01PAF, The Swedish Research Council: research grant K2006-74X-20141-01-3 and grants from The Radiumhemmet Research Funds (JH; Stockholm group); Programme Hospitalier de Recherche Clinique Régional d’Ile de France grant AP-HP AOR 01 091 (Marie-Francoise Avril); Research grants and scholarships of the Australian National Health and Medical Research Council (G J M and R F K), The Cancer Councils of Australia, the Australian Cancer Research Foundation, the Melanoma Foundation and Cancer Research Fund of the University of Sydney (Sydney group). P A K was supported by a National Cancer Institute Preventive Oncology Academic Award (K07 CA80700). Some data collection for this publication was assisted by the Utah Cancer Registry supported by NIH Contract NO1-PC-35141, SEER Program, with additional support from the Utah Department of Health and the University of Utah. Partial support for all datasets within the Utah Population Database (UPDB) was provided by the University of Utah Huntsman Cancer Institute.
-
Competing interests: None.
-
Authors from Lund Melanoma Study Group:
Lund Cancer Center Department of Oncology, University Hospital, Lund, Sweden (Christian Ingvar, Ake Borg, Johan Westerdahl, Anna Masback, Hakan Olsson)
Additional authors from GenoMEL listed by group/centre:
Barcelona: Dermatology Department (Josep Malvehy) and Genetics Service (Celia Badenas, Remedios Cervera), Melanoma Unit, Hospital Clínic de Barcelona, IDIBAPS, Barcelona, Spain; Dermatology Department, Hospital Arnau de Vilanova, Universitat de Lleida, Lleida, Spain (Rosa Martí); Genetic Counseling of Cancer, Hospital Sant Joan, Universitat Rovira i Virgili, Reus, Spain and ICO, Hospital Trueta, Girona, Spain (Joan Brunet-Vidal); Boston: Massachusetts General Hospital, Boston, MA (Guang Yang); Brisbane: Queensland Institute of Medical Research (Nicholas Martin, David Whiteman, Adele Green) and Queensland Cancer Fund (Joanne Aitken), Brisbane, Australia; Emilia-Romagna: Dermatology Unit, Maurizio Bufalini Hospital, Cesena, Italy (Paola Minghetti); Genoa: Dipartimento di Oncologia, Biologia e Genetica, Universita degli Studi di Genova, Vle Benedetto XV, 6, 16132 Genova, Italy (Michela Mantelli, Lorenza Pastorino, Sabina Nasti, Sara Gargiulo), Istituto Nazionale per la Ricerca sul Cancro (IST), Largo Rosanna Benzi, 10, 16132 Genova, Italy (Sara Gliori); Leeds: Genetic Epidemiology Division, Cancer Research UK Clinical Centre, Leeds (Sushila Mistry, Juliette Randerson-Moor); Leiden: Leiden University Medical Center (Femke A de Snoo, Jeanet AC ter Huurne, Jasper van der Rhee, Leny van Mourik, Frans van Nieuwpoort), STOET, The Netherlands Foundation for the Detection of Hereditary Tumors (Clasine van der Drift), Leiden, The Netherlands; Paris: Service de Génétique, Institut Gustave Roussy, Villejuif (Brigitte Bressac-de Paillerets), AP-HP-Hopital Cochin, Université Réné Descartes, Paris (Marie-Francoise Avril), French Hereditary Melanoma Study Group: Drs F Grange, B Sassolas, F Boitier, J Chevrant-Breton, C Lasset, C Dugast, P Vabres, France; Philadelphia: Department of Genetics (Arupa Ganguly), Department of Dermatology (Michael Ming), Department of Pathology and Laboratory Medicine (Patricia Van Belle), University of Pennsylvania, Philadelphia, PA, USA;
Stockholm: Department of Oncology-Pathology, Karolinska Institute and Karolinska University Hospital Solna, Stockholm, Sweden (Anton Platz, Suzanne Egyhazi, Rainer Tuominen, Diana Linden); Sydney: Westmead Institute for Cancer Research, Sydney, Australia (Helen Schmid);
Tel-Aviv: Department of Dermatology (Alon Scope, Felix Pavlotsky), The Susanne Levy Oncogenetics Unit (Eitan Friedman), Sheba Medical Center, Sackler Faculty of Medicine, Tel-Aviv University, Israel; Utah: Department of Dermatology and Huntsman Cancer Institute, University of Utah School of Medicine, Salt Lake City, UT (Mark Eliason).
Copyright Title 17 U.SC § 105 states that copyright protection is not available for any work of the United States Government. Since my authorship contribution was done as part of my official duties as a National Institutes of Health (NIH) employee, my work is a work of the United States Government and as such is in the public domain. If the Publisher intends to disseminate the work in foreign countries, the Publisher may secure copyright to the extent authorized under the domestic laws of those foreign countries. The copyright will be subject to a paid-up, nonexclusive, irrevocable worldwide license to the United States in the manuscript of such copyrighted work to reproduce, prepare derivative works, distribute copies to the public and perform publicly and display publicly the work, and to permit others to do so. The Publisher should not pay royalty income for work done by Federal employees as part of their official duties.
This agreement shall be governed and construed in accordance with Federal law as interpreted by the Federal courts in the District of Columbia.