Article Text

Abstract
We report an epidemiological and genetic study attempting complete ascertainment of subjects with microphthalmia, anophthalmia, and coloboma (MAC) born in Scotland during a 16 year period beginning on 1 January 1981. A total of 198 cases were confirmed giving a minimum live birth prevalence of 19 per 100 000. One hundred and twenty-two MAC cases (61.6%) from 115 different families were clinically examined and detailed pregnancy, medical, and family histories obtained. A simple, rational, and apparently robust classification of the eye phenotype was developed based on the presence or absence of a defect in closure of the optic (choroidal) fissure. A total of 85/122 (69.7%) of cases had optic fissure closure defects (OFCD), 12/122 (9.8%) had non-OFCD, and 25/122 (20.5%) had defects that were unclassifiable owing to the severity of the corneal or anterior chamber abnormality. Segregation analysis assuming single and multiple incomplete ascertainment, respectively, returned a sib recurrence risk of 6% and 10% in the whole group and 8.1% and 13.3% in the OFCD subgroup. Significant recurrence risks were found in both unilateral and bilateral disease. In four families, one parent had an OFCD, two of which were new diagnoses in asymptomatic subjects. All recurrences in first degree relatives occurred in the OFCD group with a single first cousin recurrence seen in the non-OFCD group. A total of 84/122 of the MAC cases were screened for mutations in the coding regions of PAX6, CHX10, and SIX3. No pathogenic mutations were identified in the OFCD cases. A single PAX6 homeodomain missense mutation was identified in a subject with partial aniridia that had been initially misclassified as coloboma.
- microphthalmia
- anophthalmia
- coloboma
- genetic aetiology
- MAC, microphthalmia, anophthalmia, and coloboma
- OFCD, optic fissure closure defects
- DHPLC, denaturing high performance liquid chromatography
- ONC, optic nerve coloboma
Statistics from Altmetric.com
- MAC, microphthalmia, anophthalmia, and coloboma
- OFCD, optic fissure closure defects
- DHPLC, denaturing high performance liquid chromatography
- ONC, optic nerve coloboma
Microphthalmia, anophthalmia, and coloboma (MAC) are major structural eye malformations. Anophthalmia is the complete absence of the eye, microphthalmia is a small eye usually defined in terms of corneal diameter or axial length, and coloboma is a segmental ocular defect, most commonly a “keyhole” deficiency in the iris. The best estimates of the birth prevalence of microphthalmia and anophthalmia from well maintained population based registers are 14 and 3 per 100 000 births, respectively. In the last decade there has been considerable public concern and media attention aroused by reports of apparent clusters of anophthalmia and microphthalmia and of possible links between toxic environmental exposures and these conditions. A number of potential environmental causes have been proposed although the evidence in their support is only preliminary.1
The aetiology of MAC is not well understood. A significant genetic contribution to the aetiology of non-syndromal MAC has been suggested by previously observed familial clustering of these defects2 and successful linkage analysis in a small number of families where these disorders segregate in a Mendelian fashion.3,4 However, there are no published population based data from which to quantify the relative recurrence risk in these families. An understanding of the underlying genetic causes of eye defects is necessary for the optimal management of these conditions, the counselling of the affected families, and to provide insights into the biological processes involved in eye development, which in turn may lead to new prevention strategies.
We aimed to establish a population based and well characterised cohort of children with MAC in Scotland to investigate genetic aetiology and improve estimates of recurrence risk for genetic counselling of affected families. Specific objectives were to assemble a complete as possible case register of all cases of MAC in children born from 1981-1996 and resident in Scotland at birth and to validate cases by examination of affected children, where possible. Recognising the clinical heterogeneity in presentation, we also sought to develop and apply a phenotypic classification system based on knowledge of eye development. Finally, we sought to investigate the possible role of the most promising candidate genes, the developmental control genes PAX6, CHX10, and SIX3, in the aetiology of the subgroup of MAC cases with no apparent cause by screening selected subjects for germline mutations in these genes. Homozygous loss of function mutations in PAX6 have been identified in a single infant with anophthalmia associated with severe brain abnormalities.5CHX10 mutations have been described in two families with non-syndromal microphthalmia segregating as an autosomal recessive trait.6 The SIX3 gene underlies some cases of holoprosencephaly7 with one patient having microphthalmia and coloboma without other classical features of holoprosencephaly.7
METHODS
Case ascertainment
After ethical approval was obtained from all Local Research Ethics Committees in Scotland, a national system of case ascertainment was established. All MAC cases born in Scotland between 1981 and 1996 and registered in the national congenital anomalies register, the EUROCAT (European Network of Congenital Anomalies Registers) register in Greater Glasgow, and the register of the blind and the national blind school were identified. In addition, all paediatricians, clinical medical officers, Directors of Public Health, Directors of Education, clinical geneticists, community paediatricians, paediatric pathologists, ophthalmologists, and ophthalmic prosthetic departments in Scotland and the national patient support group were contacted with a request to them to notify cases to us. The completeness of case ascertainment was estimated by log linear modelling (capture recapture technique).The decision to include a case in the study was on the basis of a confirmed primary ophthalmic diagnosis of MAC by an experienced ophthalmologist.
With the consent of the parents and/or child, each case was reviewed by the study ophthalmologist and a full ophthalmic (including slit lamp and ultrasound examinations) and dysmorphological assessment (including a series of photographs and clinical measurements) was carried out. Clinical findings were reviewed by consultants in ophthalmology and clinical genetics. In cases in whom examination was not possible, the medical records of the child were reviewed. We found the available clinical classification systems of structural eye defects to be unsatisfactory as they were based on physical size of the eye with no indication of specific developmental pathology. We classified each affected eye according to whether a “6 o'clock” coloboma (iris, retinal, or optic nerve) was present. The study therefore assembled the most carefully documented population based cohort of MAC cases currently in existence.
Calculation of segregation ratios
First degree relatives were examined by an experienced ophthalmologist. Where this was not possible, medical records were reviewed to abstract relevant data. The remaining phenotypic classifications relied on family report of normal eye structure and vision. Of the 122 study cases, eight cases (seven families) were excluded from the segregation analysis: one adopted child (case No 324), four cases with recognisable patterns of malformation (three CHARGE association (case Nos 35, 40, and 96, two were concordant MZ twins), Schwartz-Jampel syndrome with nanophthalmia only (case No 60)), and three cases with known chromosome anomalies (47,XX,+18 [case 15], 46,XX,del(22)(q11.22) [case 55], 46,XY,del(5) (q15;q22) [case 45]). The remaining data set consisted of 116 affected subjects in 108 pedigrees. One of these 116 cases (case No 109) was the offspring of consanguineous (first cousin) parents. Two different segregation analyses were performed, (1) assuming single incomplete ascertainment8 and (2) assuming multiple incomplete analysis.9,10 For detailed discussion of the limitations of both approaches see Smith.11
Sample collection and mutation analysis
After written consent was obtained, DNA was prepared from samples collected either by venepuncture after application of local anaesthetic cream (preferred method) or exfoliated buccal cells (where permission for venepuncture was not given). Purified genomic DNA was used as a template to amplify the exonic sequences (including intron-exon boundaries) containing the open reading frame of PAX6, CHX10, and SIX3 using primers and PCR conditions detailed in table 1. Following amplification, mutation analysis was carried out using denaturing high performance liquid chromatography (DHPLC).12 If a shifted DHPLC peak was evident, the original DNA sample was reamplified and sequenced. For PAX6, DHPLC detected 100% of the known mutations, including a variety of mutational events such as small deletions or insertions and single nucleotide substitutions.
PAX6, CHX10, and SIX3 primers and PCR conditions
RESULTS
A total of 198 children were identified as having been born with MAC between 1981 and 1996. The diagnosis was confirmed by direct examination in 122 cases and by review of medical records in 76 cases. The birth prevalence of MAC, 19/100 000 live births, falls within the published estimates from EUROCAT registers throughout Europe and did not increase over the period 1981-1996. Capture-recapture modelling suggested the completeness of ascertainment to be 61%, so the birth prevalence could be as high as 32/100 000 (unpublished data); 39% of registrations on the national congenital anomalies register were found to be misclassified.
Classification of eye defects (table 2)
Cohort summary
A total of 85/122 (69.7%) of the examined cases had evidence of an optic fissure closure defect (OFCD) in the form of an iris, retina, or optic nerve coloboma (fig 1A). Twelve of 122 (9.8%) of cases had clinical evidence that the structural eye defect did not involve an OFCD (termed non-OFCD), but could be ascribed to primary lens (fig 1B), vitreous, or choroidoretinal pathology. Twenty-five of 122 (20.5%) cases had an unclassifiable defect, for example, a small eye with sclerocornea (fig 1C). Forty-six of 85 (54.1%) of the OFCD cases had bilateral eye defects. In 37/46 of these cases both eyes had OFCD, in nine cases one eye had OFCD with the other eye having an unclassifiable defect. Six of 12 non-OFCD cases were bilaterally affected; of these 5/6 had bilateral non-OFCD and 1/6 had one eye non-OFCD with the fellow eye having an unclassifiable defect. There were no cases in which one eye had a non-OFCD and the other eye was OFCD. Nine of 25 of unclassifiable cases were bilaterally affected. Eight families were identified with familial recurrence of a structural eye defect. In seven of these, the proband had OFCD. In all confirmed cases, the other affected members of these pedigrees had OFCD with no non-OFCD eyes identified. In the one family where the proband had a unilateral non-OFCD, the recurrence was in a first cousin who also had unilateral non-OFCD (fig 2H). These pedigrees are presented in detail below.

Examples of eyes with OFCD (A), non-OFCD (B), and unclassifiable (C) structural eye defects.
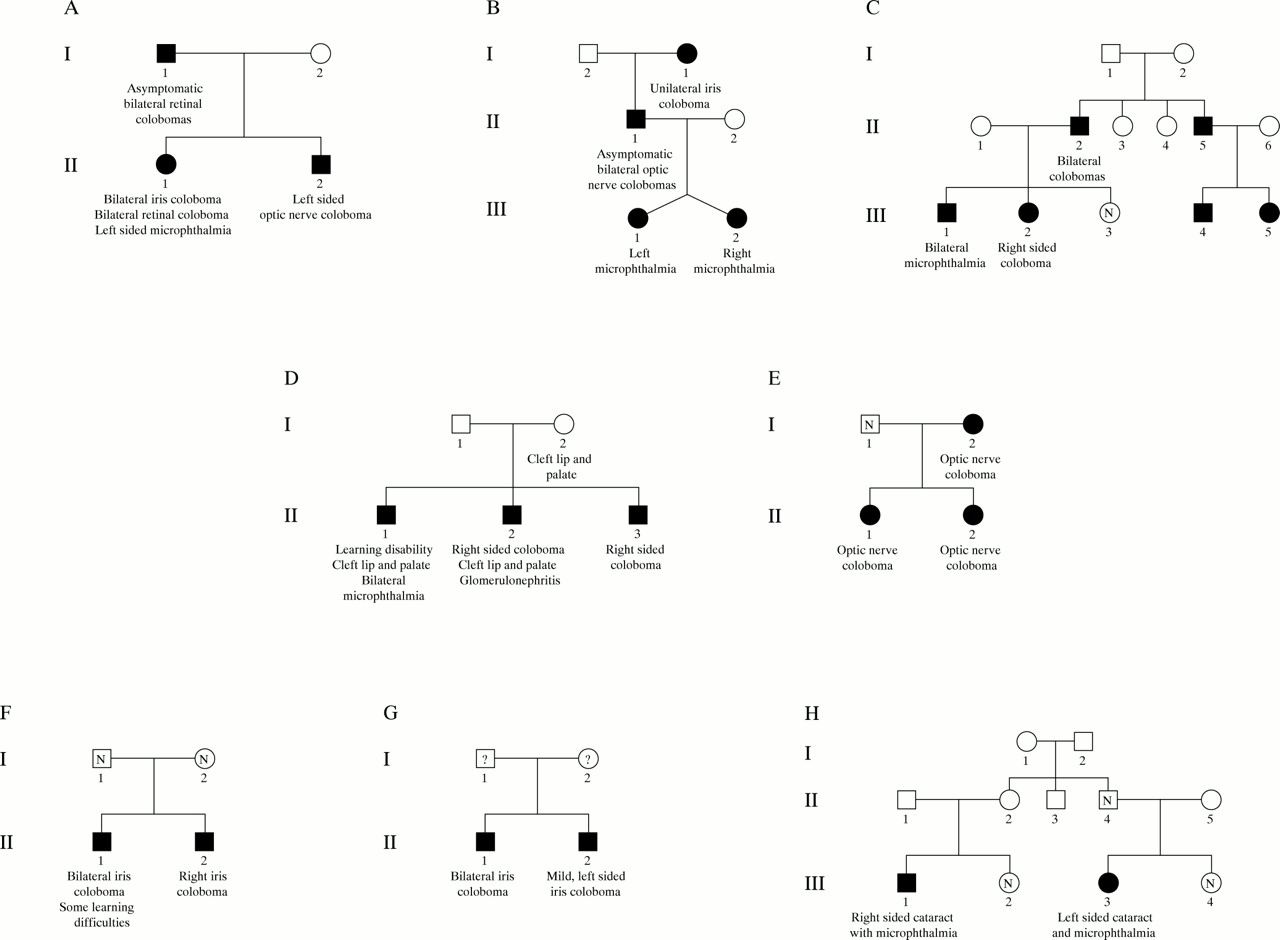
Pedigrees of the eight multiplex families identified in the study.
Sib recurrence risks
Table 3 gives details of the estimation of the segregation ratios assuming single incomplete and multiple incomplete ascertainment (and confidence intervals) for all cases of MAC and for the subgroups with OFCD. Details of the families with recurrences are given in fig 2. Families 1-3 (fig 2A-C) are consistent with autosomal dominant inheritance of a structural eye defect. . Family 4 (fig 2D) appear to have a previously described autosomal dominant condition, coloboma, uveal, with cleft lip and palate and mental retardation (OMIM 120433).13 Family 5 (fig 2E) is consistent with either autosomal or X linked dominant inheritance.13 It is not possible to determine accurately the inheritance pattern in families 6-8 (fig 2F-H). Family 8 (fig 2H) shows the only recurrence observed in a non-OFCD family. The calculated γs using the sib recurrence risks for the whole group (0.06-0.10) and our observed birth prevalence (19/100 000) was 316-527.14
Calculated segregation ratios. Sib recurrence risk derived from segregation analysis
Extraocular phenotypes
Forty of 122 cases had one or more associated major malformations. Seven of 40 were the cases mentioned above that were excluded from the recurrence risk analysis and 2/40 were from family 5. The remaining 31 cases were very phenotypically heterogeneous with few representing “clean” syndrome diagnoses. These cases are detailed in table 4. Twenty-six of 122 cases had learning disability.
Extraocular phenotypes
Molecular analysis
Mutation analysis of PAX6, CHX10, and SIX3 showed no unequivocally pathogenic sequence alterations. A previously undescribed single nucleotide substitution (1087G→C) resulting in a missense mutation in the homeobox of PAX6 was detected. This substitutes a highly conserved arginine residue at position 242 of the PAX6 protein by threonine (R242T). Arginine 242 is located in the middle of the second alpha helix of the paired type homeodomain. The role of this helix is to stabilise the third helix, which makes sequence specific contacts with the DNA. This mutation has not been detected in over 160 other subjects analysed. Apparent heterozygosity for this mutation was detected in a child with what was initially thought to be a mild 6 o'clock iris coloboma of the left eye (fig 3). A subsequent examination by a consultant paediatric ophthalmologist (BF) showed that the defect did not actually extend beyond the anterior layer of the iris and was thus an atypical partial aniridia. There is no family history of congenital eye malformation and the right eye of the child was completely normal. This missense mutation was subsequently identified in blood DNA from his phenotypically normal mother. A number of shifted peaks on DHPLC were detected in both PAX6 and CHX10, but upon sequencing these were found to be intronic or silent coding region polymorphisms, which were present at a similar frequency in control DNA samples.

{kind=link}
{kind=link}
{kind=link}
Partial aniridia in the left eye of male child with R242T mutation in PAX6. The right eye was completely normal.
DISCUSSION
This study shows the limitations of relying solely on data from the national registration of congenital anomalies for the purposes of epidemiological analysis. There were relatively poor levels of completeness and accuracy for MAC reporting compared to the confirmed data from our study. Even using multiple diverse sources of ascertainment we are likely to have identified fewer than 2/3 of all cases in Scotland. Certainly, some of the malformations that we examined were very mild and did not cause visual disability. It is likely that some of these cases have not presented to any medical service and therefore would be missed by our approach.
Our aim in embarking on this study was to provide a basis for genetic and epidemiological studies of MAC. We were initially impeded by the impracticality and lack of rationale of available methods for categorisation of these defects. We devised a simple clinical classification system for structural eye malformations. The presence of a coloboma was taken as implying failure of closure of the optic fissure. The optic fissure forms by apposition of the inferior lips of the optic cup and fuses at Carnegie stage 16 to form the primitive globe. An excellent description of this process is provided by O'Rahilly and Muller.15 Thus, cases were classified as: optic fissure closure defect (OFCD), not OFCD, unclassifiable (for example, anophthalmia), or unknown (for example, relative with affected eye but unavailable for further examination).
We hypothesised (a priori) that a robust classification system would show concordance of the defect type in bilaterally affected subjects and in family recurrences within classification groups, a similar spectrum of associated abnormalities and syndrome diagnoses within classification groups and a different spectrum between classification groups. We considered that such a classification system would imply the existence of aetiologically distinct groups, would improve genetic counselling, epidemiological analysis of malformation clusters, and possible teratogenic effects, and would enable a more rational approach to candidate gene analysis within groups. The observed data were consistent with our a priori hypotheses detailed above. In particular, there was consistency of defect type in paired ocular findings and familial recurrence patterns showed no discordance. This basic phenotypic classification system should help standardise reporting of these defects internationally. It would appear to aid the provision of accurate genetic counselling to individual families and may also facilitate future pooling of recurrence risk estimates from different populations. Finally, it may aid the selection and evaluation of candidate genes.
The calculation of empirical recurrence risks for sibs of affected subjects was a primary aim of our project. This has proved challenging because of the extraordinary range of phenotypes found in the cohort and the difficulty of deciding which cases to exclude from the analysis. We chose to exclude very few cases. This decision was informed by one of our index cases (family 5) who had a complex phenotype with associated malformations and learning disability while one of his two affected sibs had isolated coloboma. In support of this approach, when all cases with associated extraocular malformations were excluded (table 1B), the recurrence risk was only slightly lower than the whole OFCD group.
All the sib and offspring recurrences we observed were in the OFCD group. The recurrence risk was 33% higher in those with bilateral disease. In bilateral cases if both parents had a normal eye examination this risk dropped substantially. Some of the affected parents were asymptomatic. We suggest that a significant proportion of bilateral OFCD malformations are autosomal dominantly inherited and that detailed ophthalmological assessment of parents should be performed before giving genetic advice. A significant recurrence risk was also observed in unilateral OFCD cases. In none of these families was a parent affected, which may suggest the action of a mutation of lower penetrance in these families. Interestingly, in all these families the proband had an affected right eye. This observation, if confirmed, perhaps implies that the mutation may affect a gene that is asymmetrically expressed in early embryos and is critical for eye development. PITX2 would be an example of such a gene.16 The unclassifiable group have a lower recurrence risk than the OFCD group. This is surprising as these represent the most severe eye defects and may represent new mutations in eye development genes. This may become clear when follow up shows the offspring recurrence risk in this group. All these recurrence risks are substantially higher than those currently accepted and used for counselling purposes.
There was a high incidence of associated extraocular malformations and learning disability in this cohort. A few cases had features of recognised associations or syndromes with previously undescribed additional malformations (table 4). It is likely that several new syndrome diagnoses are yet to be delineated in this group. This is likely to require pooling of data from many different well ascertained cohorts owing to the rarity of these striking phenotypes. Data from the EUROCAT registry have suggested that 16% of microphthalmia and anophthalmia cases have chromosomal abnormalities. This figure is considerably higher than the present study because we excluded cases of trisomy 13 (40% of subjects with trisomy 13 have structural eye defects). Thirteen percent of cases recorded in EUROCAT have other major malformations. Our figure may be higher than this because all our cases had a clinical examination in addition to a case note review.
Mutation analysis results in this study were disappointing. No definitively pathogenic mutations were identified in our candidate genes PAX6, CHX10, and SIX3. The R242T PAX6 mutation identified in a single case with partial aniridia presenting as a pseudo-coloboma would appear to be of low penetrance since the other eye in this subject was entirely normal and his phenotypically normal mother was found to carry the same mutation. An extensive search for CHX10 mutations in human microphthalmia patients has recently shown different homozygous missense mutations in the same amino acid residue in two different consanguineous families making CHX10 the first identified human microphthalmia gene.6 However, such recessive CHX10 mutations appear to account for a very small proportion of microphthalmia cases, since these two mutations were identified among 300 cases and we found no evidence of these mutations in this population based cohort study.
In family 1, the phenotype of dominant optic nerve coloboma (ONC) could be consistent with a manifestation of the renal-coloboma syndrome (MIM 12330). This is an autosomal dominant trait characterised by ONC, vesicoureteric reflux, and renal anomalies because of heterozygous mutations in PAX2. Partial mutation analysis indicated no evidence of PAX2 mutations in this family (K Devriendt, personal communication). This is consistent with a report in which a large panel of patients with a coloboma and/or microphthalmia was tested for PAX2 mutations. In that study, no mutations were found except in a subject with kidney defects consistent with a diagnosis of renal-coloboma syndrome.17
Future research
Defining the genes and developmental pathways involved in MAC would appear to be a reasonable approach to understanding the aetiology of these distressing anomalies. Such information would help to clarify the role of environmental factors in the aetiology of MAC since the role of these factors will probably be more easily identified and understood in this context. A clearer understanding of environmental factors should in turn provide the best hope of preventing MAC. In addition, a knowledge of genetic factors may allow genetic analysis and prenatal diagnosis to be offered to affected families.
We are confident that there is large genetic component in the aetiology of MAC, although in many families there is clearly reduced penetrance and extremely variable expressivity. The latter observation is underlined by the frequent finding of variable laterality with different phenotypes in the left and right eye of individual children. These observations may be explained by one or more of the following factors at play: interaction with modifier genes, interaction with environmental factors, and/or the involvement of stochastic factors in the developmental process. Priority should be given to studies in which close attention is given to phenotype definition and classification (preferably adopting a developmentally based system, as in this study) and in which multicentre collaboration is used to maximise the efficient use of international expertise in genetic analysis.
Linkage analysis approaches
Although the findings of this study showed a higher genetic contribution to the aetiology of these defects than had previously been recognised, the considerable genetic as well as phenotypic heterogeneity makes it very challenging to identify the genes involved. Careful thought needs to be given to the choice of research strategy to be adopted. Some of these families are sufficiently large to support a linkage analysis approach. Such approaches to the discovery of genes causing MAC should therefore be given renewed priority and a common (international) resource of familial cases of MAC should be assembled. However, our own unsuccessful attempt to recruit a sufficient number of family members from family 3 (fig 2C) to enable linkage analysis shows that this can be difficult.
Candidate gene approaches
New candidate genes continue to be proposed including SIX6, HES1, RX, and the EYA genes emerging from work on mammalian eye development genes considered likely to be implicated in human eye disease. However, the selection of candidate genes for further research investment remains problematical as there are no well validated criteria for their selection. With respect to MAC there are a number of known examples where microphthalmia is seen in a mouse mutant, but reduced eye size is not observed when the orthologous gene is mutated in man (for example, Mitf, the HLH gene mutated at the original mouse microphthalmia locus, which in man is associated with Waardenburg syndrome). This, coupled with the large number of potential candidate genes that could contribute to the aetiology of MAC, the labour intensive nature of mutation analysis, and the relative lack of success in identifying the key candidates argues against mutation screening of multiple candidate genes being the primary approach.
However, it may be possible to carry out an efficient and effective future strategy for candidate gene analysis based on the establishment of a well characterised set of sporadic and familial cases as a resource for an international collaborative approach by a number of genetic laboratories with recognised expertise in specific candidate genes.
Acknowledgments
This study was funded by a grant from the Scottish Executive Chief Scientists Office. Supplementary funding was provided by the Wellcome Trust, the MRC, and the European Science Foundation. Anne Williamson provided expert secretarial assistance. The MACS parents support organisation have been consistently supportive. We would like to express our deepest thanks to the children and their parents who participated in this study.