Article Text

Abstract
CONTEXT Chromosomal abnormalities that involve the proximal region of chromosome 15q occur relatively frequently in the human population. However, interstitial triplications involving one 15 homologue are very rare with three cases reported to date.
OBJECTIVE To provide a detailed molecular characterisation of four additional patients with interstitial triplications of chromosome 15q11-q14.
DESIGN Molecular analyses were performed using DNA markers and probes specific for the 15q11-q14 region.
SETTING Molecular cytogenetics laboratory at the University of Chicago.
SUBJECTS Four patients with mild to severe mental retardation and features of Prader-Willi syndrome (PWS) or Angelman syndrome (AS) were referred for molecular cytogenetic analysis following identification of a suspected duplication/triplication of chromosome 15q11-q14 by routine cytogenetic analysis.
MAIN OUTCOME MEASURES Fluorescence in situ hybridisation (FISH) was performed to determine the type of chromosomal abnormality present, the extent of the abnormal region, and the orientation of the extra chromosomal segments. Molecular polymorphism analysis was performed to determine the parental origin of the abnormality. Methylation and northern blot analyses of theSNRPN gene were performed to determine the effect of extra copies of the SNRPN gene on its methylation pattern and expression.
RESULTS Fluorescence in situ hybridisation (FISH) using probes within and flanking the Prader-Willi/Angelman syndrome critical region indicated that all patients carried an intrachromosomal triplication of proximal 15q11-q14 in one of the two chromosome 15 homologues (trip(15)). In all patients the orientation of the triplicated segments was normal-inverted-normal, suggesting that a common mechanism of rearrangement may have been involved. Microsatellite analysis showed the parental origin of the trip(15) to be maternal in three cases and paternal in one case. The paternal triplication patient had features similar to PWS, one maternal triplication patient had features similar to AS, and the other two maternal triplication patients had non-specific findings including hypotonia and mental retardation. Methylation analysis at exon 1 of theSNRPN locus showed increased dosage of either the paternal or maternal bands in the paternal or maternal triplication patients, respectively, suggesting that the methylation pattern shows a dose dependent increase that correlates with the parental origin of the triplication. In addition, the expression ofSNRPN was analysed by northern blotting and expression levels were consistent with dosage and parental origin of the triplication.
CONCLUSIONS These four additional cases of trip(15) will provide additional information towards understanding the phenotypic effects of this abnormality and aid in understanding the mechanism of formation of other chromosome 15 rearrangements.
- chromosome 15 triplication
- Prader-Willi syndrome
- Angelman syndrome
- autism
Statistics from Altmetric.com
The proximal long arm of human chromosome 15 is frequently involved in molecular rearrangements including deletions,1duplications,2 triplications,3-5translocations,6 and inversions,7 as well as in the formation of supernumerary marker chromosomes.8 ,9Approximately 70% of patients with Prader-Willi syndrome (PWS) or Angelman syndrome (AS) can be characterised by the presence of a de novo interstitial deletion of 15q11-q13 spanning approximately 4 Mb.10 ,11 This recurring deletion is one of the most common observed in humans and identifies at least three hotspots for chromosome breakage in the 15q11-q13 region, referred to as BP1, BP2, and BP3.12-16 Whether this deletion results in PWS or AS depends upon the origin of the affected homologue (paternal or maternal, respectively). This parent of origin effect is the result of the presence of oppositely imprinted gene(s) within the 15q11-q13 region.17
Interstitial duplications of 15q11-q13 of maternal origin have been identified in patients with developmental delay/learning difficulties2 and patients with autistic behaviour.18-20 Subjects with paternally derived duplications have a normal phenotype instead.2 ,19 The duplications frequently cover the entire PWS/AS critical region.2 ,11 ,19 ,21 The distal extent of two maternal duplications has been mapped between S12 and S24, the site of BP3, while the proximal breakpoint is at BP2.21 The data from these two cases would suggest that the interstitial duplications may be reciprocal products of the interstitial deletion events.21
Interstitial triplication of 15q11-q13 has been previously reported in only three patients. Triplications of either maternal or paternal origin present with an abnormal phenotype and significant developmental delay as the most common phenotypic features.3-5 The distal extent of two triplication cases has been determined to lie distal to S1043, while the proximal extent is unknown.11
In addition to interstitial triplications, tetrasomy of the PWS/AS critical region is observed in patients with a large supernumerary pseudodicentric chromosome 15, commonly referred to as an “inverted duplication of chromosome 15” (inv dup(15)). Inv dup(15) chromosomes occur with a frequency of 1/5000 live births22 and are classified into two major groups, small inv dup(15) chromosomes, which do not contain the PWS/AS critical region,8 ,15 and large inv dup(15) chromosomes, that usually contain two extra copies of the PWS/AS critical region.8 While the small inv dup(15) chromosomes are usually associated with a normal phenotype, patients with the large inv dup(15) chromosomes present with mental retardation, abnormal electroencephalogram (EEG), epilepsy, hypotonia, poor motor coordination, and other dysmorphic features.23 ,24 The proximal breakpoint regions (BP1 and BP2) found in PWS/AS patients are also involved in the formation of the small inv dup(15) chromosomes.15 The distal boundary of the large inv dup(15) chromosomes apparently involves four different breakpoints, one equivalent to the common deletion breakpoint region of the PWS/AS patients (BP3), one located between GABRB3and D15S12, and the other two distal to D15S1043.9 ,11
Here we describe four patients with an interstitial triplication of 15q11-q14 associated with developmental delay. In each case we examined the extent, the orientation, and the parental origin of the triplicated segment and the effects on methylation and expression of theSNRPN gene.
Case reports
The clinical findings associated with these patients are summarised in table 1 and are compared to other cases with molecularly characterised triplications.
Summary of patients with intrachromosomal 15q11-q14 triplication
PATIENT 1
The clinical history of this girl has been previously described (patient 2) by Pettigrew et al.25 She showed many features of the Prader-Willi syndrome such as mental retardation, obesity, compulsive eating, small hands and feet, and short stature. The external genitalia appeared normal. Menarche occurred at the age of 12 with irregular periods.
PATIENT 2
This girl was previously reported by Clayton-Smithet al.26 Briefly, she had language delay with poor vocalisation and no recognisable words at 2 years of age. She could say some single words at the age of 4 years. At 6 years of age she showed global developmental delay functioning at a 3 year old level. On examination, she had a wide based, ataxic gait and difficulty with gross motor coordination. These clinical features are usually seen to a more severe degree in Angelman syndrome. An EEG and CT scan were both normal.
PATIENT 3
This boy was born to a healthy, 25 year old mother and 28 year old father who were non-consanguineous and also had a normal daughter. Decreased fetal movements were noticed during pregnancy. Vaginal delivery at 40 weeks was uncomplicated with a birth weight of 3500 g and length of 51 cm. Apgar scores were 8 and 9 after one and five minutes, respectively. From infancy, he had generalised hypotonia and delayed milestones, smiling at six months and standing at 16 months. Eye examination at 2 months showed poor visual tracking, strabismus, and a normal retina. EEG and brain MRI at 2 months were normal. At 4 months, detailed audiological examinations performed because of non-responsiveness to acoustic stimuli were normal. General and neurological examination was otherwise unremarkable.
PATIENT 4
This girl is the daughter of two healthy, non-consanguineous parents. Pregnancy and delivery, at 40 weeks, were uncomplicated with a birth weight of 3400 g. Developmental milestones were globally delayed with walking at 20 months and use of single words by 22 months. Clinical examination at 22 months showed length 82 cm (10th centile), weight 9.75 kg (3rd centile), OFC 45.3 cm (3rd centile), ICD 2.8 cm (75th centile), palpebral fissure length 2.4 cm (25th-50th centile), ear height 4.8 cm (25th-50th centile), hand length 9 cm (3rd centile). She had brachycephaly, a round face with a short forehead and mild frontal bossing, extension of the temporal hairline to the lateral eyebrows, mild synophrys, bilateral inner epicanthic folds, slightly upward slanting palpebral fissures, myopia, strabismus, broad nasal bridge and upturned nares, slightly small jaw, normal position of the ears with midhelical hypoplasia, mild hypertrichosis of the back, mild swelling of the dorsa of both hands and feet, and slightly tapering fingers. Neurological examination showed only mild hypotonia.
Materials and methods
CYTOGENETICS AND FISH ANALYSIS
Cytogenetic analysis was performed on peripheral blood lymphocyte cultures from patients 3 and 4 and their parents using standard techniques. Preparations were GTG stained. For analysis of patients 1 and 2 see Pettigrew et al 25 and Clayton-Smith et al.26
Lymphoblastoid cell lines were established for each patient and chromosome preparations were made from these cells using standard methods. The probes used in these studies are listed in order from proximal to distal where P1=P1 clone, b=BAC, p=PAC, and c=cosmid: P1-5022 (D15S18), c512 (D15S543), p151G14 (ZNF127), D15S11, p158H23 (UBE3A), b150L13 (D15S931), b72P22 (D15S1019), b53C6 (D15S165), b184N23 (D15S144). All BACs, PACs, and P1 clones were acquired from Genome Systems Inc (St Louis, MO). p158H23 (UBE3A) was kindly provided by Dr James S Sutcliffe. D15S11 was acquired as a commercially available FISH probe (Oncor, now Vysis Inc, Downer's Grove, IL). DNA from genomic clones was isolated using an AutoGen 740 (Integrated Separation System, Natick, MA). Each clone was labelled by nick translation and hybridised as previously described.27 FISH slides were analysed using a Zeiss Axiophot microscope with filters for separate detection of DAPI, FITC, and rhodamine as well as a triple bandpass filter (No 83000, Chroma Technology, Brattleboro, VT) to detect signals simultaneously. Images were collected and merged using a cooled CCD camera (KAF 1400, Photometrics, Tucson, AZ) and IP Lab Spectrum (Signal Analytics, Vienna, VA) or Quips mFISH software (Vysis, Downer's Grove, IL). The relative positions of the probes are shown in fig1.10 ,15 ,16 ,28

Schematic representation of the physical map of 15q11-q14 (not to scale) showing the relative positions of probes and primers used in FISH and PCR analyses.10 ,15 ,16 ,28 BP1, BP2, and BP3 are the three hotspots for chromosomal deletion; the breakpoints found between S1019 and S165 and between S165 and S144 are indicated as BP4 and BP5, respectively.
MOLECULAR POLYMORPHISM ANALYSIS
Genomic DNA of the patients and their parents was prepared either directly from peripheral blood lymphocytes (parents) or from Epstein-Barr virus transformed lymphoblastoid cell lines (patients). Nine short tandem repeat polymorphic markers (STRs) inside the PWS/AS region were used to determine the parental origin of the triplicated chromosome, as previously described.10 The locations of the polymorphic markers used in this analysis are shown in fig 1. Polymerase chain reaction (PCR) assays were carried out using published methods.29 PCR products were separated on a 6% polyacrylamide/urea gel and visualised by autoradiography.
METHYLATION ANALYSIS
SNRPN exon 1 methylation analysis was performed using a methylation specific PCR assay (M-PCR), described previously,30 with the addition of 0.15 μl of α-[32P]dCTP/sample. Radiolabelled PCR products were separated on an 8% acrylamide gel and maternal (174 bp) and paternal (100 bp) bands were quantified using a phosphorimager with ImageQuaNT software (Molecular Dynamics, Sunnyvale, CA, USA). The ratio of the area under the curve of the maternal band relative to that of the paternal band and vice versa was determined for a control subject with a normal karyotype and for the four patients. The values obtained for the patients were then compared to the normal control.
NORTHERN ANALYSIS
Total RNA was purified using TRIzol Reagent from Life Technologies Inc (Rockville, MD). Twenty μg of total RNA were loaded in each lane for northern blot analysis. A cDNA probe for theSNRPN region exon –1 to exon 1 (RN175, RN140) was generated as previously described.31 The PCR product was cloned using a pGEM-T Easy vector (Promega Madison, WI, USA) and was 32P-labelled by random priming.32 Hybridisation was performed using ExpressHyb Solution (Clontech, Palo Alto, CA, USA) at 65°C. The filters were washed in a solution of 0.2 × SSC, 0.1% SDS at 65°C. An oligonucleotide specific for 28S ribosomal RNA (5′-AACGATCAGAGTAGTGGTATTTCACC -3′) was directly32P-labelled and used to correct for RNA loading differences.33 Relative intensities of the hybridised bands were quantified using ImageQuaNT software.
Results
EXTENT OF THE TRIPLICATED SEGMENTS
G banding analysis of patient 1 and patient 2 was previously reported and originally interpreted as indicating interstitial duplications.25 ,26 G banding analysis of patient 3 and patient 4 showed a 46,XX,15q+ karyotype that was also initially interpreted as probable duplication of the 15q11-q13 region (data not shown).
Dual colour FISH analysis using combinations of clones P1-5022, p158H23, b72P22, b53C6, or b184N23 was performed on interphase nuclei and metaphase chromosomes. The locations of these clones within chromosome 15q11-q14 are presented in fig 1. Patient 1 (fig 2A-C) and patient 3 showed identical results, including three signals on one chromosome 15 homologue for probes P1-5022, p158H23, and b72P22, and a single signal for clones b53C6 and b184N23. This indicates that these patients have triplications extending from a site proximal of S18 to a point between S1019 and S165 (fig 1). Patient 4 (fig 2D-F) showed three signals for clones P1-5022, p158H23, and b72P22 as well as for clone b53C6, while only clone b184N23 was present in single copy. This larger triplication, therefore, includes both S18 and S165 (fig 1). These results indicate that in these three patients the triplicated region extends from the most proximal breakpoint, BP1, to a distal breakpoint that lies between D15S1019 and D15S165 in patients 1 and 3 (BP4), and S15S165 and D15S144 in patient 4 (BP5) (fig 1).

FISH showing normal and triplicated signals on chromosome 15. (A, B, C) Metaphase cells and interphase nuclei from patient 1 showing that the triplication extends from BP1 to BP4; the abnormal 15 (arrow) has three copies of P1-5022 (D15S18, red) and of p158H23 (UBE3A, green) (A) and three copies of p158H23 (green) and b72P22 (D15S1019, red) (B). Probes b53C6 (D15S165, green) and b184N23 (D15S144, red) are both present in single copies (C). (D, E, F) Metaphase cells and interphase nuclei from patient 4 showing that the triplication extends from BP1 to BP5; the abnormal 15 (arrow) shows three copies of P1-5022 (D15S18, red) and of p158H23 (UBE3A, green) (D), three copies of p158H23 (green) and of b72P22 (D15S1019, red) (E), and three copies of b53C6 (D15S165, green) but only one copy of b184N23 (D15S144, red) (F).
FISH analysis using the same clones on interphase nuclei and metaphase chromosomes of patient 2 also showed three signals using clones p158H23, b72P22, and b53C6, indicating that the distal extent of the triplication in patient 2 is the same as that in patient 4 (BP5) (fig 1). The proximal breakpoint in this patient, however, involves a previously unobserved breakpoint that was mapped using clones c512, p151G14, and D15S11. These results indicate that in patient 2 the proximal breakpoint lies between ZNF127 and D15S11 (fig 1).
ORIENTATION OF THE TRIPLICATED SEGMENTS
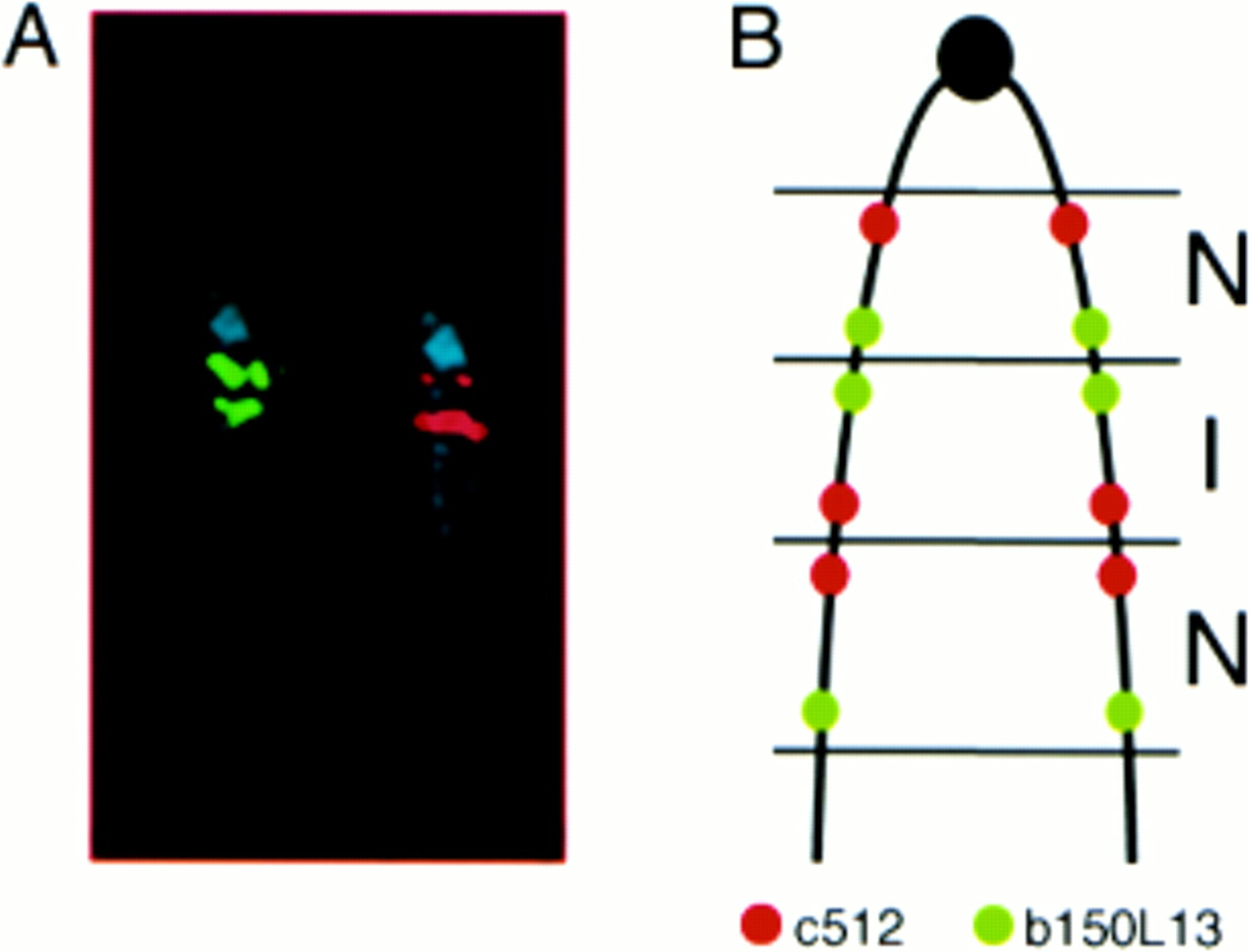
In all four patients, the orientation of the triplication was determined using two probes within the triplicated region, one located near the proximal boundary and one located ∼4 Mb further distal. The proximal probes were c512 for patients 1, 3, and 4, and D15S11 for patient 2. b150L13 was used as distal probe in all four patients. The positions of the probes used are shown in fig 1. The results showed that the distances between the signals were not equal across the triplication (fig 3A): for c512 and D15S11 the middle signal was closer to the more distal signal than to the proximal, whereas for b150L13 the middle signal was closer to the proximal signal. These results indicate that the middle repeat is inverted in orientation with respect to the two flanking segments that are normally orientated. An ideogram of the abnormal chromosome with the orientation of the repeats is shown in fig3B.

Orientation of the triplicated segments. Hybridisation with probes c512 (red) and b150L13 (green) to metaphase chromosomes of patient 4 (A) and ideogram of the triplicated chromosome 15 showing the relative position of the probes used (B). For probe c512 (red) the telomeric signal was of double intensity as compared to the centromeric signal; for probe b150L13 (green) the centromeric signal was stronger than the telomeric signal, indicating that the repeats of the triplication are normal (N) in the proximal and distal regions and inverted (I) in the central region. The same results were obtained in patients 1, 2, and 3. Note that in patient 2, c512 was replaced by D15S11 as proximal probe.
PARENTAL ORIGIN OF THE TRIPLICATIONS
Molecular analysis of microsatellite polymorphisms within 15q11-q13 showed that patient 1 has three distinct alleles for S1035, S13, S1365, and GABRB3 polymorphisms. Analysis at locus S1035 shows that the patient inherited one allele from her mother and two alleles from her father, one of which is present in two copies, as shown by the higher intensity of allele 4 in the patient compared to the same allele in her father (fig 4A). At least three different markers were tested in the other patients: D15S113, GABRB3, and GABRA5 in patient 2; D15S18, D15S11, D15S13, D15S113, GABRB3, and D15S97 in patient 3; and D15S18, D15S1035, D15S11, D15S13, D15S113, GABRB3, and D15S97 in patient 4. For each informative marker, the patients inherited one paternal and two different maternal alleles, with one of the maternal alleles being present in two copies. An example is shown in fig 4B, where allele 3 of patient 4 is twice the intensity as the same allele in the mother.

Microsatellite analysis at locus D15S1035 in family of patient (Pt) 1 (A) and at locus D15S542 in family of Pt 4 (B). The triplication is of paternal origin in patient 1 as determined by the presence of three paternal alleles and one maternal allele. Note that the intensity of allele 4 is higher than the same allele in the father, indicating that in patient 1 allele 4 is duplicated. In patient 4, the triplication is of maternal origin as indicated by the presence of three maternal alleles and one paternal allele. The increase of intensity of allele 3 in patient 4 indicates that this is the duplicated allele.
These results indicate that for patient 1 the triplication is paternal in origin, while for patients 2-4 the triplications are maternal in origin.
SNRPN METHYLATION AND EXPRESSION
Methylation in exon 1 of SNRPN was examined in all patients using methylation specific PCR (fig 5). The maternal allele gives rise to a 174 bp band and the paternal allele to a 100 bp band. A control subject with a normal karyotype was analysed to calculate the pat/mat and the mat/pat ratios in order to normalise the results obtained for our patients. DNA from patient 1 (paternal triplication) shows a paternal band approximately three times more intense than the maternal band, while in the three maternal triplication patients (2-4), the signal from the maternal allele is about three times more intense than that from the paternal allele. These findings confirm the presence of one copy of the maternal allele and three copies of the paternal alleles in patient 1, and of one copy of the paternal allele and three copies of the maternal alleles in patients 2-4.

Methylation analysis of the CpG island at the 5′ end of the SNRPN gene. In each lane the upper band (174 bp) is from the maternal chromosome and the lower band (100 bp) is from the paternal one. The Angelman syndrome patient AS with a deletion of the maternal 15q11-q13 chromosome shows only the paternal band, while the Prader-Willi patient PWS with a paternal deletion of the same region shows only the maternal band. Patient (Pt) 1 has a more intense paternal band that indicates paternal inheritance of the triplication (P/M=3.3), while Pt 2, 3, and 4 show a more intense maternal band suggesting that the triplication is of maternal origin (M/P=2.6 in Pt 2, 2.8 in Pt 3, 3.0 in Pt 4). In the normal control, the intensity of the two bands is almost the same and it is used as a reference value. Further details can be found in the Methods section.
Normally, the SNRPN gene is expressed only from the paternal chromosome.17 In order to determine if the extra copies of the 15q11-q14 region influence the expression of this paternally expressed gene, the RNA from our patients was tested by northern blotting and compared to that from a normal control. As expected, SNRPN expression increased only in the patient who inherited the paternal triplication. In the three maternal triplications, the levels were similar to those of the normal control (fig 6). A low level of expression was observed in the PWS control consistent with published data that indicatedSNRPN is 96-100% methylated from the maternal allele.34

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of the SNRPN gene. (A) Autoradiograph of a gel loaded with 20 μg of total RNA extracted from lymphoblastoid cell lines from each patient and hybridised sequentially with a cDNA probe for SNRPN and with an oligonucleotide specific for 28S rRNA to correct for RNA loading differences. (B) Relative SNRPN expression for each sample. Only in patient 1 (Pt 1), who has a paternal triplication, is the SNRPN expression more than two times increased compared to the normal controls. Patients 2, 3, and 4 (Pt 2, Pt 3, Pt 4) with a triplication of maternal origin have SNRPN RNA levels similar to the normal controls. SNRPN RNA levels were not different from the normal controls in a patient with Angelman syndrome (AS) with a deletion of the maternal 15q11-q13 chromosome, and almost absent in a patient with Prader-Willi syndrome (PWS) with a deletion of the same region on the paternal chromosome.
Discussion
This work reports the clinical and molecular findings for four subjects with de novo interstitial triplications of the 15q11-q14 region. The clinical manifestations of the four patients are summarised in table 1 and compared with the other three known cases of triplication of the same region.
Molecular analysis shows that in patients 2, 3, and 4 the triplication is maternal in origin. A maternal triplication in a case with mild features of AS was previously described by Schinzelet al.4 More recently, Longet al 5 reported a patient with developmental delay who was found to have a small supernumerary inv dup(15) and an interstitial triplication of proximal 15q11-q13 maternal in origin. The absence of euchromatin from the PWS/AS critical region within the inv dup(15) suggests that the trip(15) is responsible for that patient's abnormal phenotype. Moreover, all the de novo supernumerary marker 15 chromosomes that include the PWS/AS critical region are maternal in origin8 ,24 ,35 ,36 and are associated with moderate to severe mental retardation.8 ,22 ,24 ,37 These observations indicate that maternal rearrangements of the 15q11-q13/q14 region can be associated with an abnormal phenotype.
A paternal triplication of the 15q11-q13 region was previously found by Cassidy et al 3 in a hypotonic, developmentally delayed child with non-specific findings. Patient 1, described here, has a triplication of the 15q11-q14 region on the paternal chromosome that is associated with mental retardation, obesity, and cleft palate. These results indicate that paternal triplication of the PWS/AS critical region may also have phenotypic effects, unlike duplications that show a normal phenotype.2 ,19
By using the three markers D15S1019, D15S165, and D15S144, we have shown that the triplication involves two breakpoints located between D15S1019 and D15S165 (BP4) and between D15S165 and D15S144 (BP5) that are likely to represent the breakpoints for most large inv dup(15) chromosomes.11 Both BP4 and BP5 contain portions of the genomic duplication (duplicon) present in BP2 and BP3A/3B16 (unpublished data) that are likely to be hotspots for recombination.
Intrachromosomal triplications of chromosome 15 seem to result from a common mechanism as indicated by the inverted orientation of the middle repeat found in our patients and in previously reported cases.4 ,5 Wang et al 38 recently reviewed the published reports since 1993 and identified at least 11 cases of intrachromosomal triplication, affecting chromosomes 2q, 5p, 7p, 9p, and 15q, involving an inverted orientation of the middle repeat in all cases investigated. These authors proposed a mechanism of two U type exchanges involving three chromatids, as previously described inDrosophila by Slizynska.39 For chromosome 15, this would include a U type exchange in meiosis I between homologues at the distal breakpoint region and a second U type exchange at the proximal breakpoint region between sister chromatids of a single homologue (see fig 4 in Wang et al 38). This mechanism gives rise to a small inv dup(15), as was seen in the case of Long et al,5 but may more often be lost, and a chromosome 15 which contains three sequentially arranged segments of which the first and third repeats are in direct orientation while the second one is inverted. Another possible mechanism of recombination was previously described by Schinzel et al,4where the authors proposed that the first step in the formation of the triplication involves an unstable dicentric inv dup(15)(pter→q13::q13→pter) chromosome of maternal origin that contains both a direct and an inverted repeat. Triplication would be the result of recombination between this broken chromosome and the remaining normal chromosome 15 followed by the loss of the supernumerary marker.
The phenotypic abnormalities associated with the chromosomal rearrangements of the 15q11-q14 region can be determined by alterations in the expression of the imprinted genes mapping within the PWS/AS critical region. At least five imprinted, paternally expressed genes have been localised to the centromeric end of the PWS/AS critical region, including ZNF127,40 NDN,41 MAGEL2,42 SNURF/SNRPN,43 ,44 and IPW.45 PAR-1 andPAR-5 are paternally expressed sequences which remain poorly characterised.46 UBE3A is preferentially expressed from the maternal chromosome in brain and is localised distally to the paternally expressed cluster of genes.47 The region also includes a cluster of three GABAA receptors with unclear imprinting status.48 ,49
In each patient we studied the methylation status of the CpG island located upstream of the SNRPNgene.46 Our results showed that the presence of the 15q11-q14 triplicated region does not interfere withSNRPN methylation status, maintaining hypomethylation on the paternal chromosome and hypermethylation on the maternal chromosome. Accordingly, SNRPN RNA levels increased only in the patient with paternal triplication (patient 1), while in the three maternal triplication patients,SNRPN expression was normal. These results suggest that an alteration in the levels ofSNRPN RNA may play a role in the pathogenesis of the phenotype of patient 1. However, the expression of other imprinted and non-imprinted genes mapping in the 15q11-q14 region was not investigated in this study and probably also contributes to the abnormal phenotype observed in these patients.
Results presented in this paper suggest that a single, common mechanism gives rise to the proximal 15q triplications observed in our patients and additionally identify breakpoints in common with other chromosome 15 rearrangements. These data constitute an important contribution to our understanding of the nature and mechanism of formation of these chromosomal rearrangements, as well as the derivation and potential familial recurrence of AS, PWS, and pervasive developmental delay/autism. Taken together, this information may aid in providing appropriate genetic counselling, especially in the context of prenatal diagnosis.
Acknowledgments
We would like to thank Dr L B K Herzing and Dr R J Leventer for critical reviewing of the manuscript and Dr J S Sutcliffe for providing the UBE3A FISH probe. This work was supported by NIH grant HD36111.