Article Text

Abstract
Prepubertal periodontitis (PPP) is a rare and rapidly progressive disease of young children that results in destruction of the periodontal support of the primary dentition. The condition may occur as part of a recognised syndrome or may occur as an isolated finding. Both autosomal dominant and recessive forms of Mendelian transmission have been reported for PPP. We report a consanguineous Jordanian family with four members affected by PPP in two nuclear sibships. The parents of the affected subjects are first cousins. We have localised a gene of major effect for PPP in this kindred (Zmax=3.55 for D11S901 at θ=0.00) to a 14 cM genetic interval on chromosome 11q14 flanked by D11S916 and D11S1367. This PPP candidate interval overlaps the region of chromosome 11q14 that contains the cathepsin C gene responsible for Papillon-Lefèvre and Haim-Munk syndromes. Sequence analysis of the cathepsin C gene from PPP affected subjects from this Jordanian family indicated that all were homozygous for a missense mutation (1040A→G) that changes a tyrosine to a cysteine. All four parents were heterozygous carriers of this Tyr347Cys cathepsin C mutation. None of the family members who were heterozygous carriers for this mutation showed any clinical findings of PPP. None of the 50 controls tested were found to have this Tyr347Cys mutation. This is the first reported gene mutation for non-syndromic periodontitis and shows that non-syndromic PPP is an allelic variant of the type IV palmoplantar ectodermal dysplasias.
- prepubertal periodontitis
- periodontal disease
- cathepsin C
- linkage
Statistics from Altmetric.com
Although it is becoming increasingly apparent that there is a significant genetic aetiology in human susceptibility to periodontitis, to date no gene of major effect has been identified for any of the non-syndromic forms of periodontitis. Identification of such a gene would help develop the nosology of periodontal diseases and add to emerging molecular pathogenesis models of periodontal disease. Prepubertal periodontitis (PPP) is a rare and rapidly progressive form of early onset periodontitis that results in destruction of the periodontal support of the primary and secondary dentitions.1 The condition may be localised (usually to deciduous molars) or generalised to all teeth.2 3 Both autosomal dominant and recessive patterns of familial transmission have been reported.4 5 The varied clinical phenotype that has been reported for PPP probably reflects the fact that the term PPP has been used to describe an aetiologically heterogeneous group of conditions that share an overlapping clinical presentation.6 Nosology of PPP and other early onset periodontitis based upon clinical findings has not been helpful in developing a classification scheme that reflects the aetiological basis of these diseases.
In addition to occurring as an isolated clinical finding, PPP is known to be associated with a number of genetic syndromes, including chronic familial neutropenia (MIM 1627007), leucocyte adhesion deficiency type I (MIM 1169208), leucocyte adhesion deficiency type II (MIM 2662659), Chediak-Higashi syndrome (MIM 21450010), and Papillon-Lefèvre syndrome (PLS, MIM 24500011). These syndromes each have a known genetic basis and are aetiologically diverse.
The radiographic presentation of alveolar bone loss in PPP in many cases appears similar to that observed in PLS, a dermatological syndrome characterised by palmoplantar hyperkeratosis and severe early onset periodontitis. The gene for PLS has been sublocalised to a 2.8 cM interval on chromosome 11q14, and subsequently mutations of the cathepsin C gene have been identified.12-16 Although a number of associated clinical findings have been reported to occur with PLS,17 18 the similarity in radiographic presentation of the periodontitis component of PLS with PPP led us to hypothesise that PPP may be an allelic variant of PLS. To test this hypothesis, we performed a linkage study to evaluate support for localisation of a PPP gene to the PLS candidate interval. We subsequently sequenced the cathepsin C gene for mutations that could be responsible for PPP.
Materials and methods
ASCERTAINMENT AND CLASSIFICATION OF FAMILIES
Fourteen members of a consanguineous Jordanian family segregating PPP were identified through proband ascertainment of patients presenting for treatment at the Department of Paediatric Dentistry, Al-Amir Rashid Hospital Royal Medical Services, Jordan (fig 1). Informed consent was provided by all study participants before inclusion in the study. Medical and dental histories were obtained and all family members were examined for skin lesions, particularly palmoplantar keratoderma, in order to rule out syndromic types of PPP.19 All family members received clinical and radiographic oral examinations to document the presence of periodontitis. A diagnosis of PPP was determined in subjects with severe early onset periodontitis.
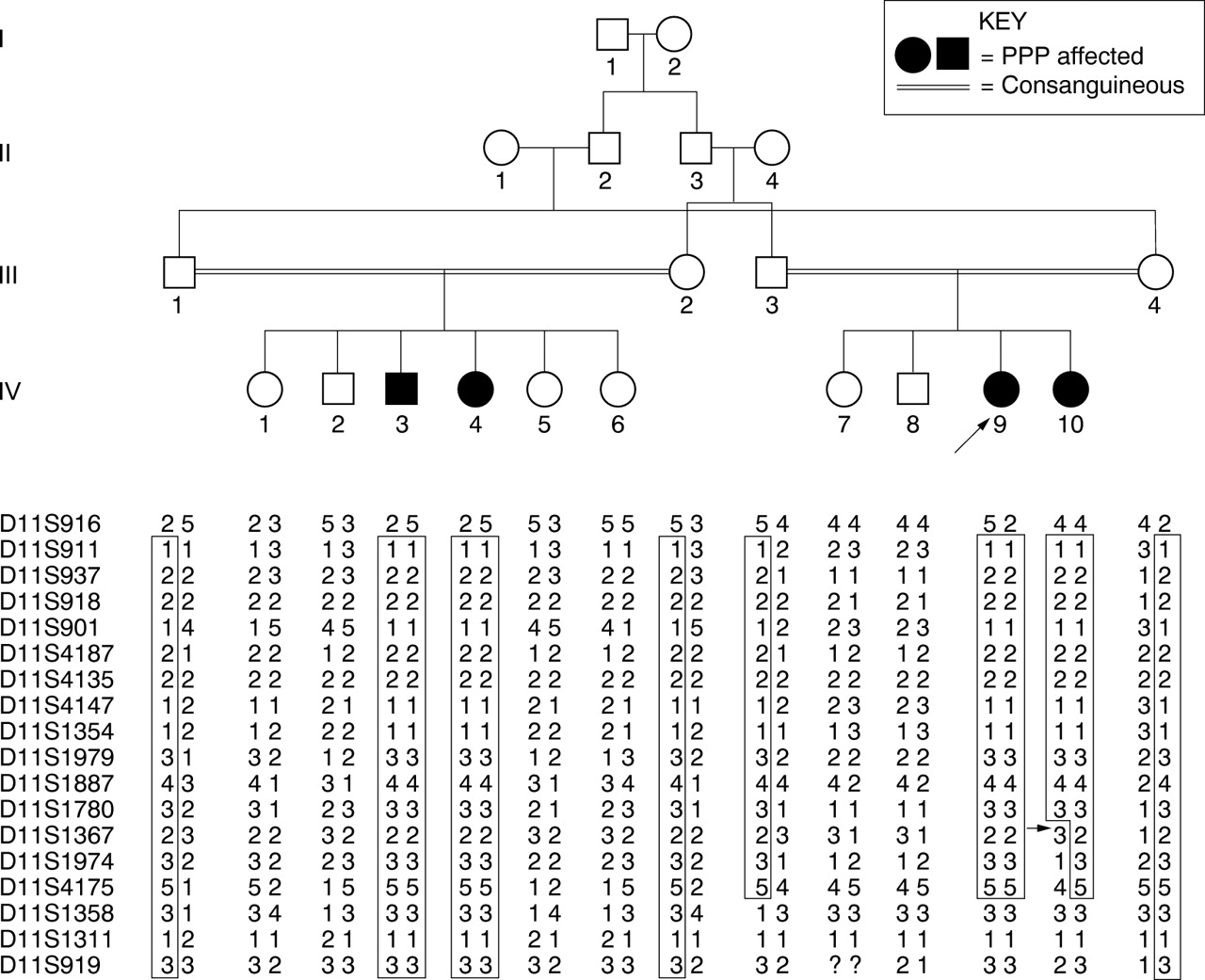
Pedigree and haplotype data of the family segregating prepubertal periodontitis (PPP). Subjects from generations III and IV were analysed for the current study. Genotypes for chromosome 11q SSLP markers spanning the PLS candidate interval were used to construct haplotypes.14 Segments which are likely to be homozygous by descent are boxed. Arrow indicates recombinant events.
DNA MARKER ANALYSIS
Peripheral venous blood (7.5 ml) was obtained by standard venepuncture. Genomic DNA was isolated using the QIAamp blood kit (Qiagen Inc). Family members were genotyped for a high density array of 18 DNA markers using standard techniques for PCR amplification with γ-32P radioactively labelled primers according to the manufacturer's protocol using a PCR 9600 thermocycler (Applied Biosystems).20 Following PCR amplification, individual samples were separated on a 6% PAGE-7 mol/l urea gel (30 W, 1500 V). Following electrophoresis, gels were wrapped in cellophane, exposed in a phosphorimaging cassette for 15 minutes, and scanned (Molecular Dynamics Inc). Alleles were scored and genotype data entered into the pedigree file of the LINKAGE computer package.
LINKAGE ANALYSIS
Linkage was assessed between PPP and 19 markers in the 11p14 region, using both parametric lod scores and model free methods. DNA markers were chosen based on an existing genetic map21 and on a correlated physical and genetic map of the PLS candidate interval.15 The LINKAGE program, with recent updates to speed calculations (VITESSE and FASTLINK22 23), was used for all single- and multipoint lod score calculations. For the lod score calculations, PPP was assumed to follow autosomal recessive transmission, with an allele frequency of 0.0001 and penetrance of 95%. A model free identity by descent method was also used to assess linkage to guard against misspecifications in the genetic model for PPP. The SimIBD approach developed by Davis et al 24 simulates marker genotypes in the affected relatives conditional on the marker genotypes in the unaffected family members to determine an empirical null distribution for calculating p values. SimIBD has been shown to be robust to allele frequency misspecification. Finally, alleles at each marker were tested for association with PPP using the transmission disequilibrium test (TDT) of Cleves et al,25 as programmed in SAGE.26 Haplotype analysis was used for error elimination during the linkage scan and for the determination of the critical linkage segment. Haplotype construction was performed using the CRIMAP program with the CHROMPIC option.27
SEQUENCE ANALYSIS
The cathepsin C gene was amplified as described previously.16 28 Oligonucleotide primer sequences were designed based upon reported genomic and cDNA sequences of cathepsin C (GenBank accession numbers: full length cDNA (NM-001814) and genomic DNA of CTSC(U79415)). The PCR products were prepared for sequencing by excising appropriate bands from an agarose gel and extracting the fragments using a Qiagen Gel Clean-up Kit as previously described.16 28 The sense and antisense strands of each PCR product were directly sequenced on an ABI Prism 377 DNA Sequencer (Perkin-Elmer) using four dye terminator chemistry. Sequencing data were automatically collected and analysed by the ABI Sequence Analysis software. Raw sequence data were analysed and consensus sequences and nucleotide/amino acid alignments generated using DNASIS V2.6 for Windows (Hitachi Software Engineering Co Ltd).
Results
CLINICAL FINDINGS
Fourteen members of a consanguineous Jordanian family segregating PPP were ascertained for the study (fig 1). No family members showed signs of, nor reported a history of, palmoplantar type lesions or an increased susceptibility to infections. Of the 14 members of this extended consanguineous family who were clinically examined, four (IV.3, IV.4, IV.9, and IV.10) were found to be affected by generalised prepubertal periodontitis (fig 1). These subjects were found to have clinically evident gingival inflammation and radiographic evidence of alveolar bone loss (fig 2). No other family members were found to have periodontitis and none of the parents reported a history of early onset periodontitis.
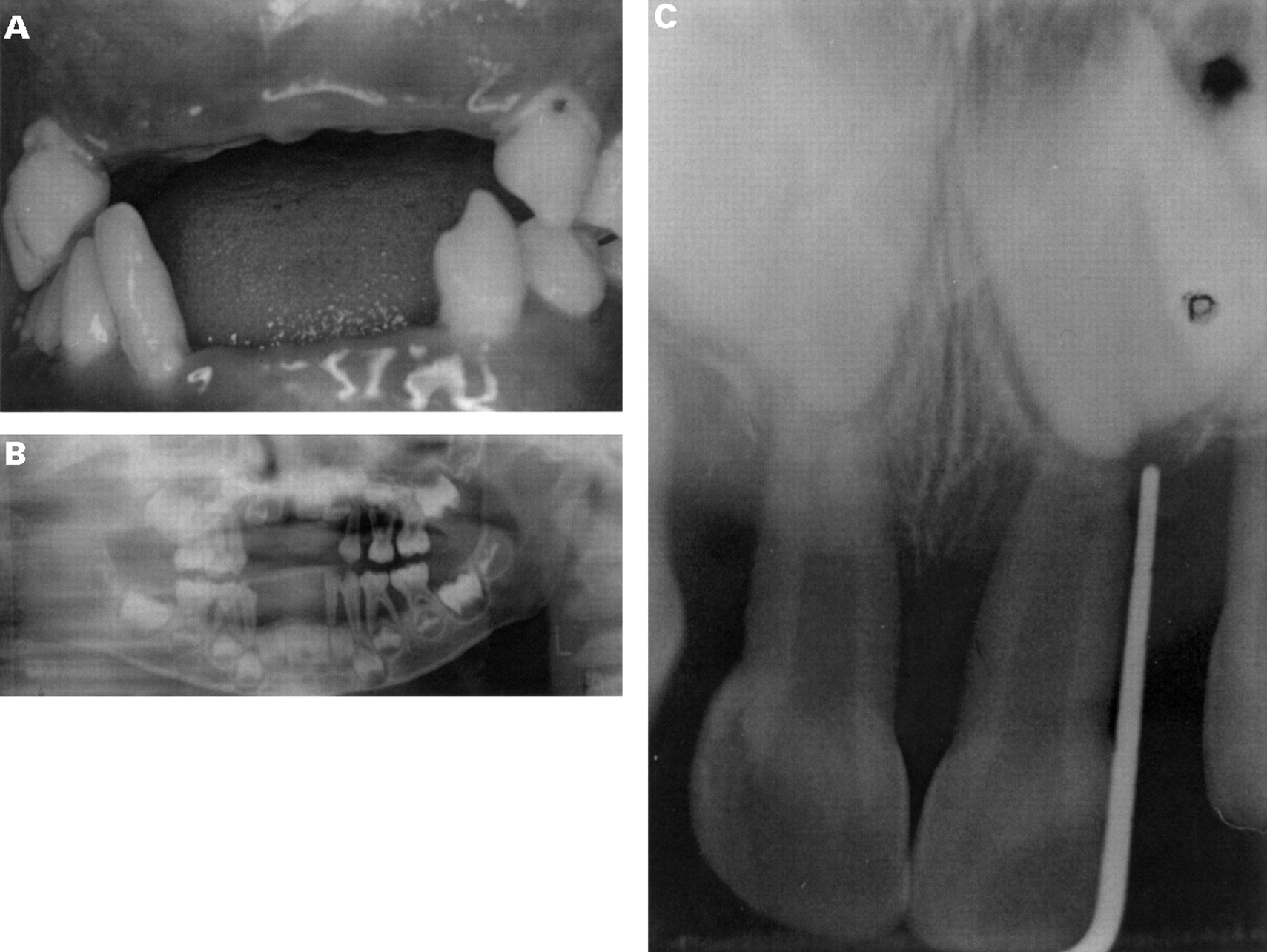
Clinical photos of IV.10 aged 4 years. (A) Photograph showing gingival recession around remaining primary dentition. All maxillary and three mandibular incisors have been prematurely exfoliated. (B) Panographic radiograph of IV.10 showing advanced alveolar bone loss around the primary dentition (teeth shown clinically in (A)). (C) Periapical radiograph showing anterior alveolar bone loss around primary central incisors in IV.9.
TWO POINT LINKAGE ANALYSIS
The results of the parametric and model free methods for assessing linkage and association between PPP and the 18 markers are summarised in table 1. Two point lod scores were consistent with a major gene for PPP in this extended family in the interval flanked by D11S916 and D11S919 (Zmax=3.55, D11S901 at θ=0.00). The results from SimIBD and TDT approaches were also consistent with linkage to this interval. Furthermore, multipoint linkage analysis yielded a maximum multipoint lod score of 4.14 at D11S1354. The results of the multipoint analysis of these DNA markers with the PPP trait are summarised in fig3.
Results of two point linkage analysis and model free tests of association (SimIBD and TDT)

In order to define the smallest interval containing the PPP locus, all subjects were analysed for recombination events by haplotype reconstruction. Haplotypes of family members for the DNA markers genotyped are shown in fig 1. All affected subjects (IV.3, IV.4, IV.9, and IV.10) were homozygous for a common haplotype spanning D11S911 to D11S1780. All four parents of the affected subjects were heterozygous carriers of this haplotype, consistent with inheritance of both maternal and paternal copies of this genetic interval from a common familial ancestor (“identical by descent”). The cosegregating segment in which recombination was not detected was flanked by the markers D11S916 and D11S1367 (fig 1). Relevant obligatory recombination events could be identified between the PPP locus and marker loci that placed the disease gene locus proximal to D11S1367 and distal to D11S916, a distance of 14 cM. This cosegregating linked region overlaps with 2.6 cM of the refined interval that contains the PLS gene locus on chromosome 11q14.15 Mutations in the cathepsin C gene have recently been identified as causing PLS.16 Correlation of physical and genetic maps of this region of chromosome 11q14 localised the cathepsin C gene within the interval segregating with the PPP phenotype.15
SEQUENCE ANALYSIS
To determine if mutations of the cathepsin C gene were responsible for PPP, the gene was sequenced in all family members. Sequence analysis showed PPP affected subjects from these families were homozygous for a 1040A→G missense mutation of the cathepsin C open reading frame (fig 4). This mutation changes the wild type TAT codon to a TGT codon, resulting in amino acid 347 changing from tyrosine to cysteine (Tyr347Cys). No other exon sequence variations were found in the affected subjects. All four parents of the affected subjects were heterozygous carriers of Tyr347Cys mutant alleles. Two of the clinically unaffected sibs (IV.1 and IV.6) were also found to be heterozygous carriers of Tyr347Cys mutant alleles. None of the heterozygous carriers was found to have current clinical evidence of early onset periodontitis nor history of past occurrence. Of the 50 controls tested, none was found to have this Tyr347Cys missense mutation or any sequence variation at this position.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sequence analysis of CTSC. The numbering of the wild type sequence is based upon the cDNA sequence of CTSC (Accession NM-001814). (A) Wild type sequence. (B) III.2 from fig 1. The arrow indicates the heterozygous sequence at nucleotide 1040, with ATG start codon as +1. (C) IV.4 from fig 1. The arrow indicates the A→ G (Y347C) missense mutation.
Discussion
Periodontal diseases can be broadly grouped into two forms, gingivitis and periodontitis. Clinically, gingivitis is associated with a reversible inflammation of the soft tissues surrounding the teeth. Periodontitis refers to a heterogeneous group of diseases that result from an irreversible destruction of the periodontal tissues (the junctional epithelium, periodontal ligament, alveolar bone, and connective tissue). Periodontitis is estimated to occur in 15-20% of the adult population and in 0.5-3 % of children, depending on the population studied.29 Although several classification schemes have been reported, the general consensus is that currently there is insufficient knowledge to separate truly different forms of periodontal diseases from differences in presentation of the same disease.1 6 30 Existing classifications of periodontal diseases based upon histology, localisation, and severity of attachment loss has not been helpful in understanding the pathomechanism of the disease.
In emerging disease paradigms, most forms of periodontal diseases are believed to be aetiologically complex, involving the interplay of both environmental and genetic risk factors. Although specific microbial (gram negative microbes) and environmental agents (for example, cigarette smoking) important in periodontal disease initiation and progression have been identified, the primary determinant of whether they cause disease appears to be determined by the nature of the host response they illicit.31 To date, the specific molecular targets of these microbial and environmental agents are unknown. As a result, molecular progression models of periodontitis pathogenesis are not well developed. While it is increasingly apparent that there is a significant genetic basis for periodontitis, it is clear that most adult forms of disease (AP) are genetically complex.
In contrast to adult forms of periodontitis, there is evidence that a gene of major effect may be important in early onset periodontitis (EOP). Numerous reports of familial aggregation of EOP are consistent with a genetic basis for the disease.32 The most comprehensive segregation analysis of the EOP, evaluating over 100 families, supported genetic transmission of the trait.33 A major gene for a localised form of juvenile periodontitis has been mapped to chromosome 4q12-21, although the gene responsible has not been identified, and this form of EOP appears genetically heterogeneous.34 35
The EOP have been broadly classified as prepubertal forms, affecting the primary dentition, and juvenile forms, affecting the permanent dentition in subjects less than 35 years of age.6 The distinction between different forms of EOP is unclear, and both prepubertal and juvenile forms of EOP can segregate in the same family.2 Prepubertal periodontitis can occur as an isolated trait (non-syndromic) and also in syndromic association with additional clinical findings.1 The genetic basis of several such syndromes is known and gene mutations have been reported for Chediak-Higashi syndrome (defective CHS polypeptide36), leucocyte adhesion deficiency type I (mutations of the lymphocyte function associated antigen 1 gene37 and diminished expression of beta 2 (CD18) integrins38), and leucocyte adhesion deficiency (LAD) type II (associated with mutations in a still unidentified GMD regulating protein39). Not all genetic defects in syndromes associated with periodontitis involve immune defects. Defective collagen has been reported in Ehlers-Danlos syndrome (types IV and VIII) suggesting that structural integrity of the periodontal tissues may also predispose to periodontitis.40 41 While the presence of periodontitis in these genetic syndromes suggests that genetic factors are determinants of susceptibility for early onset periodontitis, it is unclear whether the periodontitis is a direct result of the primary genetic defect, or rather represents a downstream consequence of the primary defect.
Localisation of a major gene for PPP to chromosome 11q14 and identification of a cathepsin C mutation in affected subjects confirms a genetic aetiology in this form of non-syndromic PPP. Generalised prepubertal periodontitis was the only clinical finding in the affected subjects studied in this report. They did not manifest any findings reported to occur in syndromic types of periodontitis. No family members were reported to have a generalised susceptibility to other infections and dermatological disease was absent outside the oral cavity. Affected subjects were homozygous for a Tyr347Cys cathepsin C mutation. Cathepsin C mutations have recently been identified as the genetic basis for Papillon-Lefèvre syndrome and Haim-Munk syndrome, allelic variants of the type IV palmoplantar ectodermal dysplasias (table 2). As a lysosomal cysteine proteinase, cathepsin C is important in intracellular degradation of proteins and appears to be a central coordinator for activation of many serine proteinases in immune/inflammatory cells.42 The Tyr347Cys missense mutation identified in this PPP family was not present in any of the 50 unaffected controls tested. Four observations support the hypothesis that this Tyr347Cys missense mutation is responsible for PPP in this extended family. First, the CTSC locus is located within the genetic interval where PPP has been localised (fig2). Second, CTSC is a highly conserved gene43 and the Tyr347Cys missense mutation has not been identified as a normal polymorphism of the gene. Segregation of the Tyr347Cys missense mutation in affected/unaffected members of this kindred is consistent with autosomal recessive inheritance from a common ancestor (fig 1). Third, a variety of otherCTSC mutations have been identified in patients with PLS and HMS,16 28 syndromes with severe early onset periodontitis. Finally, after submission of this paper, Toomes et al 44 identified the same Tyr347Cys mutation in a family with PLS. They showed that homozygous and heterozygous Tyr347Cys subjects had decreased cathepsin C activity. Table 2 lists phenotypic associations forCTSC mutations reported to date.
Phenotype correlations with CTSC mutations reported to date
Partial expression of cardinal features of the PLS phenotype has been reported previously in several populations.46 47 We have determined that a descendant from the Cochin isolate with partial expression (no periodontitis) described by Soskolneet al 47 is homozygous for aCTSC missense mutation (Q286R). All other family members homozygous for the Q286R mutation have both severe periodontitis and palmoplantar lesions.28 Variable expression of the phenotype associated with the cathepsin C mutation may reflect the influence of other genetic or environmental factors. Identical mutations can give rise to multiple different phenotypes, such as has been shown for the craniosynostoses and fibroblast growth factor receptor mutations.45 Identical mutations in cathepsin C may also give rise to different phenotypes (PLS and PPP).
Identification of a cathepsin C missense mutation in these PPP affected subjects provides direct evidence that this form of non-syndromic PPP is also an allelic variant of the type IV palmoplantar ectodermal dysplasias. This finding suggests that at least a subset of non-syndromic EOP conditions may be oral manifestations of dermatological diseases. Clearly, the generality of cathepsin C mutations in other forms of periodontitis needs to be determined. A more complete understanding of the functional physiology of cathepsin C in health and in the pathogenesis of periodontitis carries potentially broad implications for diagnosis and treatment of periodontitis.
Acknowledgments
We thank the families for participating in these studies and Peggy Bobby, Jared J Marks, and Xiaoqu Lu for technical assistance. This work was supported by National Institutes of Dental and Craniofacial Research R01-DE11601 and R01-DE12920. Some of the results of this paper were obtained using the program package SAGE, which is supported by a US Public Health Service Resource Grant (1 P41 RR03655) from the National Center for Research Resources.