Article Text

Abstract
Cryptic subtelomeric rearrangements are suspected to underlie a substantial portion of terminal chromosomal deletions. We have previously described two children, one with an unbalanced subtelomeric rearrangement resulting in deletion of 22q13→qter and duplication of 1qter, and a second with an apparently simple 22q13→qter deletion. We have examined two additional patients with deletions of 22q13→qter. In one of the new patients presented here, clinical findings were suggestive of the 22q13 deletion syndrome and FISH for 22qter was requested. Chromosome studies suggested an abnormality involving the telomere of one 22q (46,XX,?add(22)(q13.3)). FISH using Oncor D22S39 and Vysis ARSA probes confirmed a terminal deletion. A multi-telomere FISH assay showed a signal from 19qter on the deleted chromosome 22. Results were confirmed with 19qtel and 22qtel specific probes. The patient is therefore trisomic for 19qter and monosomic for 22qter. The patient's mother was found to have a translocation (19;22)(q13.42;q13.31). We also re-examined chromosomes from two patients previously diagnosed with 22q deletions who were not known to have a rearrangement using the multi-telomere assay. One of these patients was found to have a derivative chromosome 22 (der(22)t(6;22)(p25;q13)). No evidence of rearrangement was detected in the other patient. Thus we have found the 22q13 deletion to be associated with a translocation in three of four patients. This report illustrates the usefulness of examining patients with hypotonia, severe language delay, and mild facial dysmorphism for this syndrome and suggests that most of these deletions may be unbalanced subtelomeric rearrangements.
- multi-telomere FISH
- partial monosomy 22q
- 22qter deletion syndrome
- subtelomeric rearrangement
Statistics from Altmetric.com
Patients with the terminal 22q13 deletion syndrome share a number of clinical features, including severe expressive language delay, generalised hypotonia, developmental delay, and mild facial dysmorphism.1 The increasing number of patients being reported suggests that this may be yet another important form of mental retardation resulting from a small terminal deletion. Since first described in 1985, 22 cases have been reported. Eighteen cases were believed to result from de novo, simple, subtelomeric deletions,1-9 two cases were derived from balanced translocations,4 9 one case was the result of a familial chromosome 22 inversion,10 and in one case the mechanism was not determined.11 (It is possible that one of these cases may have been reported twice,1 7 in which case the total number would be 21 cases and the simple deletions would be 17 cases.) This report describes the clinical findings in a new case of 22q13→qter deletion and the identification of translocated material on the deleted chromosome using multi-telomere fluorescence in situ hybridisation (FISH).
Recent data indicate that some apparently terminal chromosome deletions are, in fact, derivative chromosomes involving cryptic terminal rearrangements.12 13 It is important to detect and identify the presence of subtelomeric material from other chromosomes in these deletions in order to establish accurate karyotype-phenotype associations as well as to provide important genetic information to the parents and the family members. One of the methods used to investigate cryptic chromosomal abnormalities in patients with unexplained mental retardation12 is multi-telomere FISH. This technique simultaneously detects all human telomeric regions on a single microscope slide using chromosome specific subtelomeric probes. We used this method to further analyse samples from patients in whom there was evidence suggesting the presence of a terminal chromosomal deletion or rearrangement. We studied three patients known to have 22q13→qter deletions in which the presence of translocated material was either not suspected or was in question. Including our previously reported case (case 1 in reference 9), three of four deletion 22q13 cases investigated were, in fact, derivative chromosomes resulting from subtle or cryptic translocations.
Case report
The proband was a 4 year old female who presented for evaluation of hypotonia, developmental delay, small stature, and microcephaly. She was the 3204 g product of a 38 year old gravida 1. She was noted to have been inactive in utero, although ultrasound and amniocentesis studies carried out because of maternal age were unremarkable. Delivery was uncomplicated. Problems in the newborn period included enlargement of the clitoris and subsequently delay in acquisition of motor milestones developed. At 9 months, she was hypotonic and was not yet sitting. She sat at 12 months and walked at 2 years. During the first year of life, there were concerns about feeding and at 12 months she was placed on a nutritional supplement, after which she gained weight.
On examination at 4 years 5 months, her head circumference was 46.5 cm (<2nd centile), height 90.9 cm (<5th centile), and weight 13.8 kg (<5th centile). She was alert and only mildly dysmorphic with mild flattening of the nasal bridge and the midface. She was very sociable and interactive. Although by history she does use sounds for four or five objects, no words were spoken. She could vocalise, point to some body parts, and follow one or two step commands. She had moderate hypotonia, but normal muscle mass and good strength. Sensation was intact. There was no ataxia. She was able to walk and rise from the floor without difficulty. Her reflexes were diminished.
Magnetic resonance imaging at 2 years showed colpocephaly, a decrease in the periventricular white matter with some delay in white matter myelination, and a cerebellar arachnoid cyst. A second MRI performed at a later age showed an increase in myelination and no progression of any abnormalities. Laboratory studies indicated normal lactic acid, plasma amino acids, and urinary organic acids. Secondary to her presentation and with our previous experience, blood was sent for chromosomal karyotype and FISH analysis for evaluation for a possible 22q terminal deletion.
Material and methods
Prometaphase chromosomes were prepared using synchronised peripheral blood lymphocytes.14 Chromosomes were G banded by Wright stain using trypsin (GTW) and analysed under the light microscope. FISH was performed using standard techniques, with rapid wash conditions (72°C, 2 × SSC) and no signal amplification. Slides were counterstained with either propidium iodide or DAPI depending on the fluorophores used. The following probes were used for analysis of the 22qter region: STS WI-941 and D22S39 (Oncor) and ARSA (Vysis). These probes were originally developed as control probes for the DiGeorge critical region probes and were commercially available for that purpose. Specific subtelomeric probes (Vysis) containing the loci D19S238E (mapped to 19qtel), MS607 (mapped to 22qtel), and a probe still under development (mapped to 6ptel) were also used to confirm the findings obtained from multi-telomere analysis.
The multi-telomere FISH assay (Cytocell Chromoprobe Multiprobe-T) was carried out on peripheral blood lymphocytes according to the manufacturer's instructions. Two μl of fixed cell suspension were applied to each square area of the 24 square multiprobe sample slides and then flooded with 3:1 methanol:acetic acid solution. Metaphase spread quality was evaluated using phase contrast and the slides were aged overnight at room temperature. The next day, the slides were incubated in 2 × SSC for two minutes at room temperature and dehydrated serially for two minutes each in 75%, 80%, and 95% cold ethanol. One μl of 37°C hybridisation solution was applied to each raised boss of the multiprobe device, prewarmed to 37°C, before the sample slides were positioned on top. Denaturation was performed at 75°C for five minutes and overnight hybridisation was carried out in a 37°C humidified chamber. Slides were washed and the signals were amplified following manufacturer's instructions. Slides were counterstained with DAPI and the signals were detected using a Zeiss Axioskop equipped with fluorescence and appropriate filter sets. Images were captured using a cooled charge coupled device camera (Sensys, Photometrics) and Vysis m-FISH Smart Capture software.
Results
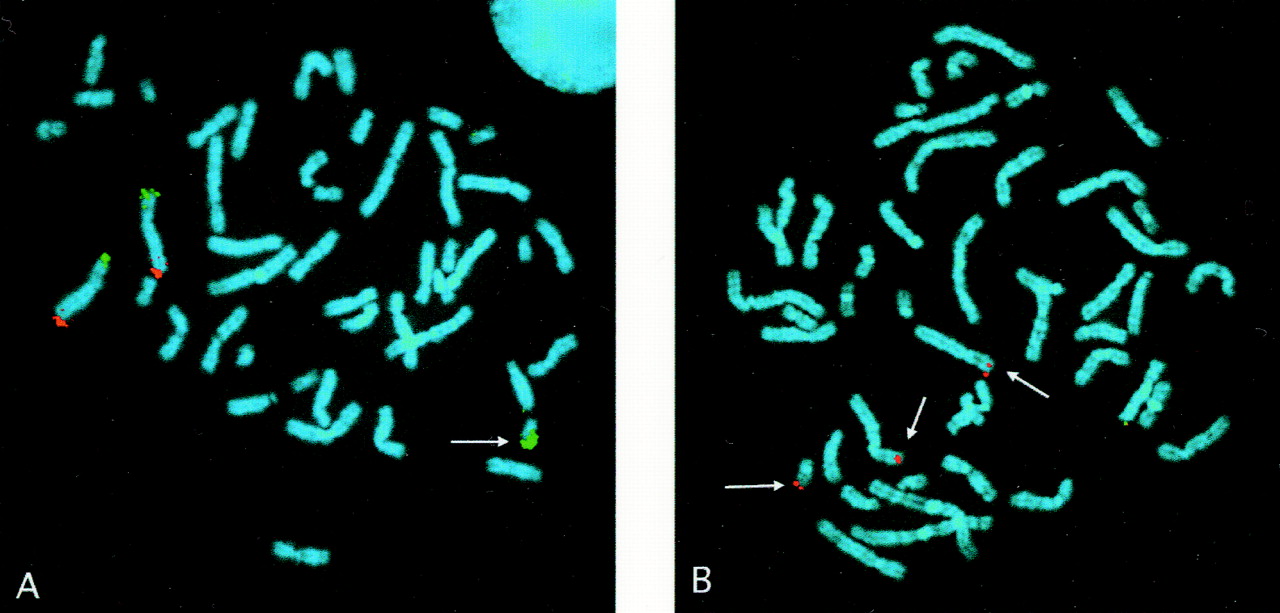
High resolution chromosome analysis (650 bands) of the proband showed a subtle abnormality at the terminal end of one chromosome 22. While a terminal deletion, rearrangement, or exchange was suspected, the abnormality was too subtle to permit the precise nature of the alteration to be determined. FISH analysis using Oncor D22S39 (not shown) and Vysis ARSA (arylsulfatase A locus) (fig 1A) probes indicated a terminal deletion of the abnormal chromosome 22, while the Oncor STS WI-941 probe (fig 1B), which hybridised more proximally than other probes, was not deleted. The breakpoint thus lies between STS WI-941 and D22S39. The 19q specific subtelomeric probe on the multi-telomere FISH assay showed three signals, with the extra signal hybridising to the distal end of the abnormal chromosome 22 (fig 1C). As expected, the chromosome 22q specific subtelomeric signal was present on only one chromosome 22 (not shown). There was no signal on the abnormal chromosome. These findings are compatible with partial trisomy for 19qter and partial monosomy for 22qter. The results were further confirmed by an individual chromosome specific subtelomeric probe for 19qter (D19S238E) (fig 1D).

FISH analysis of the patient. (A) Vysis DiGeorge probe cocktail (TUPLE1 red, ARSA green); arrow indicates deleted 22. (B) Oncor dual color DiGeorge probe cocktail (D22S75 red, WI-941 green); arrows indicate signal for WI-941 on both chromosomes 22. (C) Cytocell Multiprobe-T device, section 19 (short arm green, long arm red); arrow indicates red signal (long arm) on one chromosome 22. (D) Vysis 19qtel specific probe (red); arrows indicate signals on both chromosomes 19 and the der(22) chromosome.
High resolution G band analysis of the maternal chromosomes indicated the presence of a subtle 19qter and 22qter translocation (46,XX,t(19;22)(q13.42;q13.31)) (fig 2). This was confirmed by FISH studies using the 19qtel specific probe (D19S238E) and the 22q specific probe (ARSA) (not shown). The patient's father had normal karyotype and FISH studies (46,XY.ish 22q11.2(ARSA × 2)).

Partial karyotype of the patient's mother with ideograms. Left: der (19) and normal 19; right: der(22) and normal 22.
Following the discovery of a cryptic translocation in the above family, multi-probe FISH assays were performed on two patients previously shown in our laboratory to have terminal 22q deletions. One of these patients (case 2 described by Doheny et al 9) was believed to have a simple 22q deletion, although a derivative chromosome could not be excluded. When re-examined by multi-telomere analysis, three signals were observed using the 6p specific probe, the extra signal being present at the terminal end of the deleted 22q (fig 3A). The presence of 6p material was independently confirmed by FISH analysis with a 6p specific subtelomeric probe (fig 3B). Karyotype and FISH studies in the mother were normal. This patient's father was unavailable. The multi-telomere FISH studies of a second patient confirmed the 22qter deletion but failed to detect any evidence of a translocation, that is, two signals located on the appropriate chromosomes were seen with all other subtelomeric probes.

{kind=link}
{kind=link}
{kind=link}
FISH analysis of a previous case. (A) Cytocell Multiprobe-T device, section 6 (short arm green, long arm red); arrow indicates green signal (short arm) on the der (22). (B) Vysis 6 pter specific probe (red); arrows indicate signal on both chromosomes 6 and the der(22) chromosome.
Discussion
The increasing number of patients with terminal 22q deletions identified during genetic evaluation for unspecified developmental delay suggests that this abnormality may represent yet another important cause of unexplained mental retardation. The cases reported here, in addition to the two cases detected unexpectedly during studies for other microdeletion syndromes,2 support this assumption. Although the clinical diagnostic criteria for this syndrome have not yet been fully delineated, we feel that patients with delayed development, hypotonia, and severe expressive language delay should be examined for a deletion of terminal 22q. While high resolution chromosome studies may suggest the possibility of a terminal abnormality, our experience indicates that FISH analysis is required to confirm the presence or absence of the 22q13 deletion.
The distal 22q probes provided as controls with the commercially available DiGeorge critical region probes have been successfully used in many cases to detect this deletion. However, since these probes are not specifically designed for 22qter analysis, it is important to be aware that the distal 22q control probe differs in various commercial preparations. The more proximal Oncor probe WI-941 (no longer available for sale but possibly still being used as a control with the Oncor direct labelled dual colour DiGeorge assay) will not detect distal deletions of 22q13. On the other hand, the Oncor D22S39 probe, provided as a control for the Oncor indirect labelled DiGeorge assay, detected this deletion in all patients studied in our laboratory. Additionally, while not used for all our patients, the Vysis control probe ARSA detected the deletion in the two patients examined with this probe, while the multiprobe kit (Cytocell) detected the deletion in the three patients who were examined for the presence of cryptic translocations. Although our original patient9 was not studied with this device, there is no reason to suspect that it would not have also detected this abnormality. We suggest that while the ARSA probe packaged with the DiGeorge/velocardiofacial syndrome probe available from Vysis will detect many 22q13 deletions, more distal deletions may require one of the 22q specific subtelomeric probes that are now available.
Many terminal deletions have been found to be derivative chromosomes acquired through cryptic terminal rearrangements.4 9 12 13 15 Four cases with 22q terminal deletion4 9 (and this report) have been found to be the unbalanced products of subtle subtelomeric translocations. Although the study of one family (der(22)t(6;22)) was incomplete, the mother in the other family (der(22)t(19;22)) was found to be a balanced carrier. The information obtained by investigating the nature of the deletions in cases such as these will not only facilitate genetic counselling and prenatal diagnosis but may also result in important genetic information for immediate or extended family members.
In summary, three of four children with 22qter deletions detected in our laboratory had unbalanced subtelomeric translocations. Two of these were cryptic. We speculate that such subtelomeric rearrangements resulting in 22qter deletion may occur more frequently than has been detected. We therefore conclude that whenever 22qter deletions are identified, subtelomeric studies should be performed to characterise the abnormalities further. Our data suggest that such studies in this cohort of patients will be justified by a high yield of clinically important information for families and will also make possible more accurate phenotypic characterisation of these deletions.
Acknowledgments
We wish to acknowledge the very important contributions to this effort by Janet Biscoe, June Chung, Carol Miller, Susan Morsey, and Shirley Perdue, cytogenetic technicians in the Kennedy Krieger Institute laboratory. We also thank Dr John Proffitt and Vysis Inc for providing critical subtelomeric probes. These investigations were supported in part by the NICHD Mental Retardation Research Grant HD 24061.