Article Text

Abstract
Approximately 10% of melanoma cases report a relative affected with melanoma, and a positive family history is associated with an increased risk of developing melanoma. Although the majority of genetic alterations associated with melanoma development are somatic, the underlying presence of heritable melanoma risk genes is an important component of disease occurrence. Susceptibility for some families is due to mutation in one of the known high penetrance melanoma predisposition genes: CDKN2A, CDK4, BAP1, POT1, ACD, TERF2IP and TERT. However, despite such mutations being implicated in a combined total of approximately 50% of familial melanoma cases, the underlying genetic basis is unexplained for the remainder of high-density melanoma families. Aside from the possibility of extremely rare mutations in a few additional high penetrance genes yet to be discovered, this suggests a likely polygenic component to susceptibility, and a unique level of personal melanoma risk influenced by multiple low-risk alleles and genetic modifiers. In addition to conferring a risk of cutaneous melanoma, some ‘melanoma’ predisposition genes have been linked to other cancers, with cancer clustering observed in melanoma families at rates greater than expected by chance. The most extensively documented association is between CDKN2A germ line mutations and pancreatic cancer, and a cancer syndrome including cutaneous melanoma, uveal melanoma and mesothelioma has been proposed for BAP1 germ line mutations. Other medium to high penetrance melanoma predisposition genes have been associated with renal cell carcinoma (MITF, BAP1) and glioma (POT1). These associations between melanoma and other cancers hint at the possibility of common pathways for oncogenesis, and better knowledge of these pathways may improve understanding of the genetic basis underpinning familial melanoma. It is likely that ‘melanoma’ risk genes will impact on mutation screening and genetic counselling not only for melanoma but also a range of other cancers.
- CDKN2A
- BAP1
- familial
- melanoma
- POT1
Statistics from Altmetric.com
Introduction
The concept of melanoma risk is dynamic and multifaceted, owing to the diverse aetiology and heterogeneous nature of the disease. Genetic, phenotypic and environmental risk factors all contribute to melanoma predisposition. The majority of alterations underlying the genetic basis of this disease occur as random acquired mutations within melanocytes, and an accumulation of genomic changes contribute to melanoma development, progression and evolution. However, the presence of heritable germ line variants is an important component of melanoma susceptibility.1 Genes that predispose to melanoma are typically grouped into low, medium and high penetrance genes.2 Penetrance relates to the likelihood of a mutation carrier developing the disease over time, and reflects the overall contribution of a specific gene polymorphism, or mutation, to melanoma risk. Although no single presently known germ line alteration guarantees melanoma development, the main impact of predisposition genes is the elevation of baseline melanoma risk. For an individual with moderate to high genetic susceptibility, it is likely that fewer somatic mutations are required to accumulate before a critical level for oncogenesis is reached. Additionally, melanoma risk genes may interact directly with other genes or environmental risk factors to influence and activate melanoma growth pathways.1 ,3
A positive family history is associated with an increased risk of developing melanoma, and is particularly significant when there is a first-degree relative with multiple primary melanomas, or single primary melanomas in two or more first-degree relatives.1 The most common gene implicated in familial melanoma is cyclin-dependent kinase inhibitor 2A (CDKN2A), accounting for predisposition in approximately 20–40% of melanoma families.1 Despite a handful of other known high penetrance genes, many cases of familial melanoma are not accounted for molecularly, and the genetic basis for susceptibility remains unexplained for a large percentage of families. This suggests a likely polygenic mechanism of inheritance, including multiple low-risk alleles and genetic modifiers, as well as the possibility of rare mutations in other high-penetrance genes yet to be discovered. The risk genes that underpin familial melanoma may also be relevant to other cancers. Familial clustering of additional cancers has been observed in melanoma families, particularly pancreatic cancer linked to CDKN2A mutations, and the evidence for melanoma being part of broader cancer syndromes is mounting.1 ,4
High penetrance genes
Cyclin-dependent kinase inhibitor 2A
The CDKN2A gene on chromosome 9p21 consists of four exons that encode two unrelated proteins in different reading frames arising from alternatively spliced transcripts. p16 inhibitor of cyclin-dependent kinase 4 (p16INK4A) is produced from the α transcript of exons 1α, 2 and 3, whereas p14 alternate reading frame (p14ARF) is produced from the β transcript of exons 1β, 2 and 3. The main tumour suppressor activity of p16INK4A is through inhibition of cyclin-dependent kinases 4 and 6 (CDK4 and CDK6), thus maintaining retinoblastoma protein (RB) in a hypophosphorylated state to prevent cell cycle S-phase entry.5 p14ARF is a positive regulator of p53, and therefore a loss of p14ARF allows for accumulation of DNA damage as cells escape the senescence barrier.5 The structure of CDKN2A into two reading frames means that mutations can affect either p16INK4A, p14ARF or both, depending on which exon is affected. Autosomal dominant inheritance of germ line CDKN2A mutations has been implicated in approximately 20–40% of familial melanoma, although the mutation frequency varies between different geographical regions.5
Geographically linked founder mutations have been documented, with some occurring as a single predominant mutation based on common ancestry. CDKN2A founder mutations have been found in Sweden and the Netherlands, namely p.Arg112dup and p16-Leiden, respectively, both located in exon 2 and originating in northern Europe approximately 2000 years ago.6 Another dominant variant has been identified in Iceland, with G89D mutation contributing to the genesis of approximately 2% of all invasive cutaneous melanoma in that country.7 In Europe, G101W occurs as a founder mutation in France, Italy and Spain.5 A number of common mutations are shared between Australia and the UK, including M53I, IVS2-105A/G, R24P and L32P, reflecting a shared ancestry from British colonisation of Australia in the late 18th century.5 Differences in mutation penetrance between regions likely reflect a combination of genetics and environment associated factors, where family members are predicted to share the same ultraviolet radiation (UVR) exposures as well as a number of other heritable genetic modifiers.1 Several independent features have been associated with positive CDKN2A mutation status, including multiple primary melanomas, high number of family members with melanoma, Breslow thickness >0.4 mm and early age of melanoma onset.8 ,9 Compared with the relatively high penetrance in cohorts of familial melanoma, a much lower lifetime risk has been identified for individuals with CDKN2A mutation in population-based analysis.10
Among the high penetrance familial melanoma genes, CDKN2A is unique in that it has also been identified as a low penetrance gene conferring increased risk of melanoma in the general population. Genome-wide association studies (GWAS) have shown that variants located around the CDKN2A locus are associated with cutaneous melanoma, naevus count and tanning ability.2 ,11–13 Several independent variants are proposed to contribute to complex association signals in the CDKN2A region, and the association with melanoma risk is likely to involve multiple single nucleotide polymorphisms (SNPs).2 These SNPs include rs869330 and rs7023329 within the MTAP gene, and rs1101970 in CDKN2B-AS1 (figure 1).2 ,14
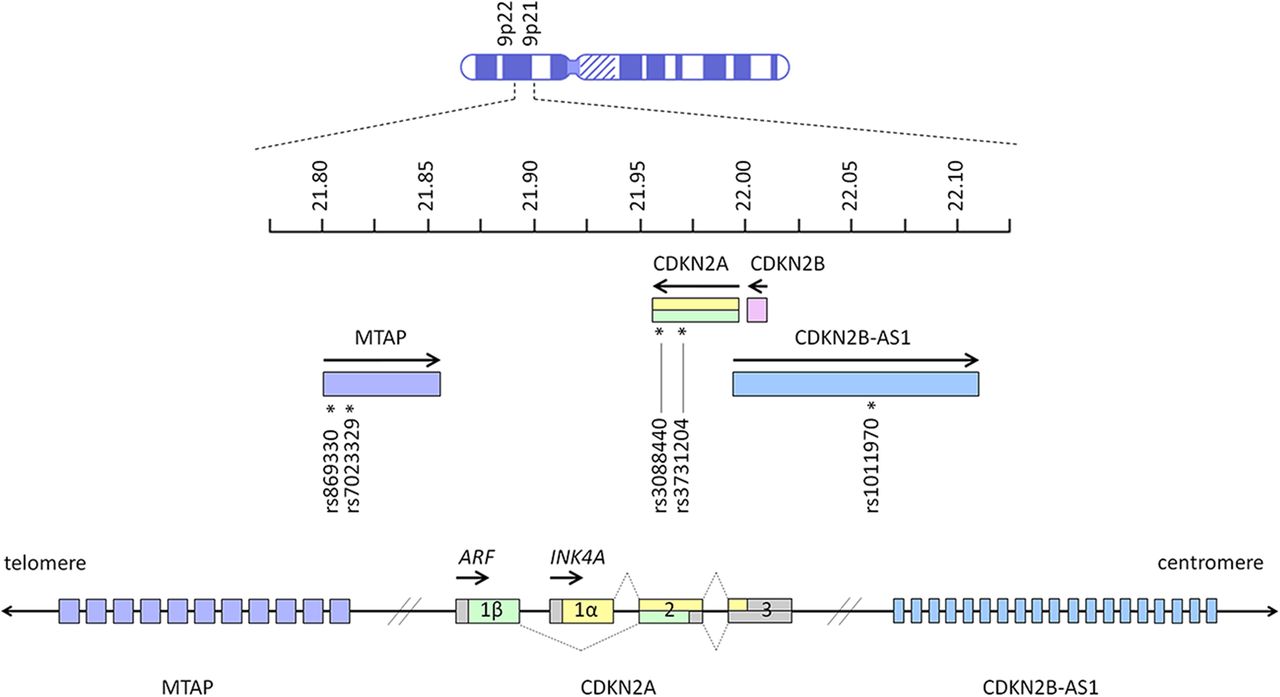
Key melanoma-associated single nucleotide polymorphisms on chromosome 9p21 and 9p22 in MTAP (rs869330 at position 21804617, and rs7023329 at position 21816528), CDKN2A (rs3088440 at position 21968159, and rs3731204 at position 21984661) and CDKN2B-AS1 (rs1011970 at position 22062134).2 ,14 Arrows indicate the direction in which genes are transcribed. Units next to the chromosome ideogram indicate megabase position of each gene from the terminus of the short arm of chromosome 9. Exons (open boxes) of CDKN2A are numbered, and dotted lines show how alternative splicing generates the alternate reading frame (ARF) and INK4A gene products.
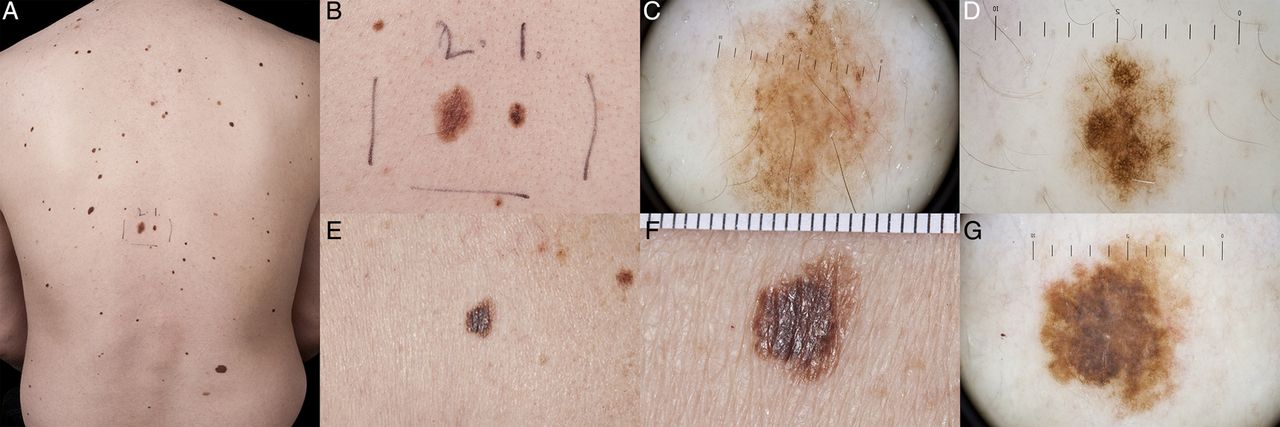
The above locus is a naevus associated region, with CDKN2A mutation carriers displaying a higher total naevi number and total naevi density compared with non-carriers.15 Phenotypic naevus differences have also been observed, with mutation carriers demonstrating significantly more clinically atypical naevi (figure 2).15 An atypical naevus has been defined as having one or more of the following clinically observed features: size >5 mm in diameter, border or contour irregularity, colour asymmetry or multiple colours, and diffusion of pigment. Some CDKN2A coding region mutation carriers have a clinical phenotype consistent with atypical naevus syndrome, historically also sometimes referred to as dysplastic naevus syndrome, however the variability of phenotypic expression means that not all carriers have atypical naevi.15

Atypical naevi showing the hallmarks of large size, border or contour irregularity, colour asymmetry or multiple colours, and diffusion of pigment. (A) many atypical naevi on the back; (B) close-up view of centre back naevi seen in A, showing highly irregular contour and colour variation; (C) dermoscopy of left naevus marked ‘2’ in (A and B), demonstrating peripheral reticular distribution of pigment relatively homogeneously associated with some centrally distributed globules and pigment reinforcement. Overall the lesion is relatively symmetrical; (D) dermoscopy of right naevus marked ‘1’ in (A and B) showing atypical reticular distribution of pigment with asymmetry in the vertical axis. Relative enlargement of pigment network in focal areas is more central with some radial streaming. Overall the lesion is relatively homogeneous in colour and does not have any blue/white veiling, regression or other hallmarks of melanoma; (E) an atypical naevus of large size, asymmetry, irregular pigmentation and contour; (F) close-up view of naevus seen in (E and G) dermoscopy of naevus seen in (E and F), showing predominantly reticular distribution of pigment with some areas of amorphous pigment. Asymmetry in the vertical axis, heterogeneous distribution of pigment and irregular borders. Multiple brown/grey dots centrally and symmetrically distributed. No blue/white veil, no regression or other hallmark of melanoma (diagnosis to be interpreted in the context of other lesions on the same patient).
Cyclin-dependent kinase 4
Germ line mutations in CDK4 on chromosome 12q14 impact the same pathway as CDKN2A mutations, and the oncogenic effects of CDK4 mutations are primarily via the control of cell cycling in the G1 phase.16 Two different mutations have been identified, in codon 24 of exon 2, leading to substitution of arginine with either histidine or cystine. These R24C and R24H mutations lead to CDK4 behaving as a dominant oncoprotein through loss of binding to p16, its negative regulator.16 Thus far, a total of 18 families with CDK4 mutations have been identified worldwide. The R24C variant has been found in six families, from France, Italy, the UK and the USA.16 The R24H variant has been found in the other 11 families, comprising three Latvian families, two French families, and one family each from Australia, Denmark, Greece, Italy, Norway and the UK.16–19 In an analysis of 17 families, median age at first melanoma diagnosis was 39 years, and the lifetime mutation penetrance based on the available data was estimated at 74%.16 The low frequency of CDK4 mutations means that very large population studies are required to accurately assess the contribution of CDK4 mutations to the overall burden of familial melanoma and the penetrance of cutaneous melanoma in the context of these mutations.
BRCA1-associated protein-1
Germ line inactivating mutations in BRCA1-associated protein-1 (BAP1), a tumour suppressor gene on chromosome 3p21, were initially identified in two distinct syndromes. Testa et al20 identified one as characterised by familial aggregation of mesothelioma and uveal melanoma, and Wiesner et al21 concurrently described the other as characterised by multiple morphologically distinct cutaneous melanocytic neoplasms and uveal melanoma. The familial aggregation of cancers associated with a proposed BAP1 syndrome has subsequently been expanded to include cutaneous melanoma, and additional neoplasms are increasingly being linked to BAP1 germ line mutations, including meningioma, cholangiocarcinoma, renal cell carcinoma (RCC) and basal cell carcinoma.22–28 The diversity of cancers suggests that the inactivating mutation is variably penetrant for different tumour types, and possibly that mutations in BAP1 depend on other unidentified genetic modifiers for a cancer phenotype to be expressed. The first recurrent BAP1 mutation has recently been reported in three families from two continents, with one family carrying a likely independent mutation based on founder haplotype analysis.28 A clustering of uveal and cutaneous melanoma in these families, and the presence of only one mesothelioma case, supports the hypothesis that specific BAP1 variants predispose to certain subsets of cancers.28
A cutaneous phenotypic feature for BAP1 germ line mutations has been proposed by the presence of multiple 0.2–1.0 cm pink to tan papules and nodules, termed ‘melanocytic BAP1-mutated atypical intradermal tumours’ (MBAITs), or alternatively, ‘BAPomas’.21 ,29–31 These lesions are similar but histopathologically distinct from atypical Spitz tumours, lacking characteristic Spitz naevi features, and are also phenotypically distinct from naevi seen in carriers of mutations in other melanoma predisposition genes such as CDKN2A (figures 2 and 3).31 As these lesions typically occur at a younger age than other cancers, accurate identification could alert to the possibility of BAP1 mutation and prompt amplified cancer surveillance. Although the MBAITs associated with BAP1 mutations were initially reported not to progress to cutaneous melanoma, atypical features of faint orange-red pigment, red papule morphology and halo formation have been described in cutaneous melanomas of individuals in BAP1 mutation positive families.23 ,24 ,32 The features may represent an overlap between the phenotype and cutaneous melanoma, either through transformation of an existing MBAIT, or de novo melanoma development with a phenotype influenced by the specific BAP1 cancer pathway. Of 21 presently reported families affected by BAP1 mutations, 16 families had at least one individual affected by cutaneous melanoma, confirming the place of melanoma in the BAP1 syndrome.20 ,22–25 ,27–29 ,31–35 BAP1 functional inactivation is also proposed to contribute to a small proportion of sporadic cutaneous melanoma, with an absence of BAP1 expression on immunohistochemistry staining described in approximately 5% of tumours.36 Therefore, in the context of familial aggregation of cutaneous melanoma, a tumour with somatic loss of 3p and/or the loss of BAP1 protein expression may suggest screening for a BAP1 germ line mutation is warranted.

Examples of melanocytic BAP1-mutated atypical intradermal tumours (MBAITs)/BAPomas, demonstrating pink to tan papules and nodules, usually symmetrical in shape and of fairly uniform colour, which contrasts with the phenotype of atypical naevi (figure 2), often seen in CDKN2A mutation carriers.
Protection of telomeres 1
Protection of telomeres 1 (POT1) contributes to the six-component protein complex of shelterin, which protects telomeres by preventing them from being mistakenly recognised as deleterious DNA breaks, regulating telomere region DNA replication, as well as telomerase recruitment and activity.37 Two recent studies have identified nine highly penetrant germ line mutations in the POT1 gene, the majority of which affect oligonucleotide/oligosaccharide-binding (OB) fold domains, which are essential for the binding of POT1 to telomeric single stranded DNA.37–39
POT1 variants appear to be highly penetrant, with one study of melanoma families from the UK, the Netherlands and Australia observing that all nine carriers developed melanoma, in addition to some individuals developing breast and small cell lung cancer.38 Melanoma associated POT1 mutations include a p.Tyr89Cys variant of the N-terminal OB domain in a five-case family, and a splice-acceptor variant between exons 17 and 18 in a six-case family.38 Two further OB fold domain mutations, p.Gln94Glu and p.Arg273Leu, were each found in a case from different families.38
A rare novel missense variant in the OB2 domain, p.Ser270Asn, was detected in all 11 cases and obligate carriers from four Italian families, with the same variant also identified in one of two affected individuals in a bilineal Italian family.39 Although all five families were apparently unrelated, the haplotype of the POT1 region was shared by all carriers, suggesting a common ancestor approximately 10 generations ago as the source of the founder mutation.39 Two further POT1 variants, p.Gln623His and p.Arg137His, were identified in another two Italian families.39 In both studies, telomeres of POT1 mutation carriers were relatively long, which has previously been identified as a risk factor for melanoma.40
Adrenocortical dysplasia protein homolog/telomeric repeat binding factor 2 interacting protein
Recently, mutations in other shelterin complex genes have been found to predispose to melanoma (figure 4). Mutations in the adrenocortical dysplasia protein homolog (ACD) and telomeric repeat binding factor 2 interacting protein (TERF2IP) genes were identified in a study of melanoma families without known genetic aetiology.38 ,39 ,41 In a cohort of melanoma families that were wild type for known predisposition genes, segregating mutations in ACD were found in four families, and another two mutations were identified that did not segregate with all melanoma cases in the families.41 A nonsense mutation in one Australian family, p.Q320X, segregated in all four cases available for genotyping, and was associated with early age at diagnosis. Another mutation, p.N249S, was identified in an Australian family and a Danish family, with a shared founder haplotype across the ACD locus. In the Australian family, with eight confirmed and four unconfirmed cases of cutaneous melanoma, the mutation segregated in all seven cases available for testing. Of five confirmed and one unconfirmed case in the Danish family, three affected family members were found to be carriers. p.Q320X and p.N249S are within the POT1 binding domain of ACD, reflecting the key role of the ACD/POT1 subunit in mediating the elongation of telomeres.41

High, medium and low penetrance genes and their chromosome band locations. Black text denotes high penetrance genes; blue text denotes medium penetrance genes; red text denotes low penetrance genes.
TERF2IP is important in the negative regulation of telomere length, by repressing homology-directed repair. A nonsense mutation and three novel missense variants have been identified. The p.Q191R was associated with onset of melanoma at 15 years and 24 years, and is predicted to disrupt the binding site for TERF2.41 This loss is proposed to prevent TERF2IP contributing to the shelterin complex. A case-control analysis of the ACD and TERF2IP mutations in sporadic melanoma cases did not identify any carriers, indicating that rare mutations are likely to be significant only in a familial context.41
Telomerase RT
Progressive shortening of telomeres with each cell division is a characteristic of normal aging, and may be hastened by exposure to harmful environmental risks such as UVR. Maintenance of telomere length is a function of telomerase, and altered telomerase regulation contributes to the limitless replicative potential of cancer cells. Telomerase RT (TERT) encodes a catalytic subunit of telomerase, and somatic TERT promoter mutations have been identified in a variety of cancers, including melanoma.42 TERT has also recently been implicated in familial melanoma following high-throughput sequencing of four affected and four non-affected individuals in a 14-case German family.43 After the region was first identified by multipoint linkage analysis, sequencing of all genes in the region revealed several novel variants, including a T>G variant in the TERT promoter.43 This germ line mutation was found in all four affected individuals, as well as one unaffected member who was only 36 years old and had multiple naevi.43 Two affected individuals developed melanoma at age 20 years and age 30 years, in addition to other cancers, suggesting that this mutation is a rare but highly penetrant melanoma risk mutation. Screening of 168 cell lines from sporadic metastatic melanoma did not find other occurrences of this novel germ line variant, although somatic recurrent UVR-signature mutations elsewhere in the TERT promoter were present in 125 of the cell lines.43
Medium penetrance genes
The relatively low frequency of high penetrance mutations suggests that a multitude of alternative germ line mutations could help explain melanoma predisposition. Medium and low penetrance alleles are more prevalent in the general population, but singularly, they are unlikely to be enough to drive oncogenesis.44 However, the complex interplay of several of these alleles may combine to raise the level of personal melanoma risk above a critical threshold. In this regard, a component of polygenic heritability has been demonstrated to underlie all sporadic cancers.44 To date, three medium penetrance genes (ie, those with variants that have ORs of disease association of between 2 and 5) predisposing to melanoma have been identified. Interestingly, all three are involved in natural variation in pigmentation (summarised below).
Melanocortin 1 receptor gene
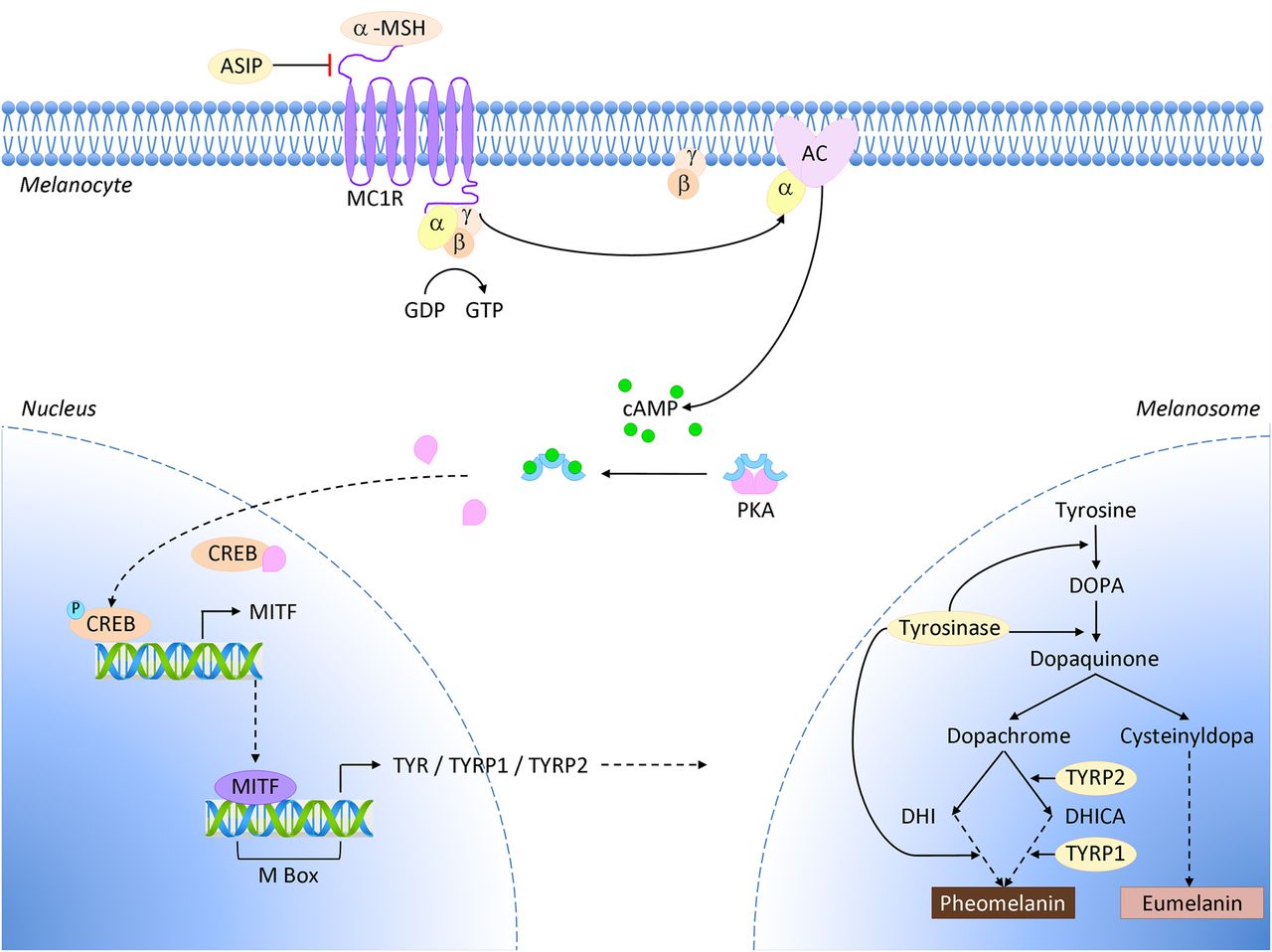
The melanocortin 1 receptor gene (MC1R) encodes the G-protein coupled receptor MC1R, which binds α-melanocyte stimulating hormone (α-MSH).45 Binding of the ligand normally activates adenylate cyclase, which then increases intracellular levels of cyclic AMP (cAMP). Raised cAMP triggers a subsequent cascade via downstream microphthalmia-associated transcription factor (MITF) and tyrosinase to stimulate melanocyte proliferation, dendricity, and eumelanin pigment synthesis (figure 5).45 The increase of photoprotective black/brown eumelanin pigments decreases the relative amount of red/yellow pheomelanins, which are poorly protective against UVR. The type and quantity of pigment determines phenotypic expression of skin and hair colour, as well as skin sensitivity to UVR and tanning response. A number of variant MC1R alleles associated with reduced cell surface receptor expression have been identified. This situation reduces binding of α-MSH, and subsequently lower cAMP levels result in less eumelanin and a greater proportion of pheomelanins.45–47

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MC1R and the pigment synthesis pathway. Binding of α-melanocyte stimulating hormone (α-MSH) to its cognate receptor MC1R on the surface of a melanocyte triggers cyclic AMP (cAMP) production via adenylate cyclase (AC). This activates the CREB and MITF transcription factors, causing an increase in expression of several components of the melanin synthesis pathway, and leads to a switch in pigment production from pheomelanin to eumelanin in melanosomes.
MC1R is highly polymorphic and a link has been established between particular alleles and a red hair colour (RHC) phenotype. Variants most strongly associated with the RHC phenotype are termed R alleles, and the consequently reduced or non-functional cell surface receptors and increased pheomelanin causes the phenotypic traits of RHC, fair skin, freckling and inability to tan.45 Other MC1R variants that are more weakly associated with RHC and have less penetrant impact on the cell receptors are designated r alleles. RHC associated MC1R variants are typically inherited in an autosomal recessive pattern.45 Variants may be inherited in a heterozygous +/R or +/r pattern, or a homozygous R/R or r/r state. Although RHC is generally a recessive trait, increases in the percentage of individuals with fair skin, blonde hair and red hair reflect in vitro studies of a dominant negative effect of MC1R variant receptors on co-inherited wild type alleles.45 Variants associated with melanoma include the R alleles D84E, R142H, R151C, R160W and D294H, (with ORs of 1.85–2.90) and the r alleles V60L, V92M, I155T, R163Q and T314T (with ORs of 1.37–2.61).48 ,49 A French case-control study recently identified 69 rare MC1R polymorphisms, including 25 novel melanoma predisposition variants.48 Just over half of the identified alleles were predicted to have a functional impact (D variants).48 Of the novel alleles, 14 D variants were exceedingly rare, each associated with only a single case in the melanoma cohort. Several others were identified in the control group.48
A pooled analysis with a large sample size from 17 case-control studies found that individuals carrying a single MC1R variant had an almost 40% increased risk of melanoma compared with homozygous wild type controls, and that the risk attributable to any MC1R variant was 28%.49 For carriers of two or more MC1R variant alleles, the risk of cutaneous melanoma was more than double the risk found for wild type controls.49 Interestingly, for individuals with the RHC phenotype, presence of MC1R variants alone was insufficient to independently predict melanoma risk.49 This could possibly reflect the significant role of environment and UVR in modulating risk for RHC variant carriers.
The analysis also revealed an association between MC1R variants and melanoma for Caucasian patients with darkly pigmented skin.49 Notably, the MC1R variant/melanoma association has been reported as stronger for photoprotective phenotypes.49 ,50 This may reflect the putative role of MC1R in non-pigment pathways, including activation of nucleotide excision repair and other DNA repair mechanisms in response to UVR damage.51–53 For variant alleles, a compromised UVR response has been attributed to a diminished α-MSH mediated oxidative stress response and reduced effects on target DNA damage response genes.53 This link to DNA repair helps explain the role of MC1R in melanoma susceptibility, suggesting that variant alleles may use either pigment or non-pigment pathways to cause melanoma. The non-pigment pathways are particularly relevant for variants that are associated with melanoma but not with the RHC phenotype.49
Microphthalmia-associated transcription factor
The MITF gene encodes the transcription factor MITF, and is a key regulator of pigment cells, including the development and differentiation of melanocytes. A recently identified recurrent germ line mutation, MITF p.E318K, is responsible for the substitution of glutamic acid at position 318 with lysine.54 ,55 The lysine residue at the site changes the binding affinity for a small-ubiquitin-like modifier (SUMO) protein, and subsequently decreases SUMOylation.54 ,55 SUMO directed post-translational modification typically impacts transcriptional regulators to inhibit transcription, and thus reduced SUMOylation effectively removes the brakes from MITF action on downstream targets.56 Comparison of expression profiles for MITF regulated targets identified 37 genes, with 17 showing modest differences in expression between wild type and p.E318K isoforms.55 This difference may indicate that variant MITF mutations have particular transcriptional affinity for specific sets of target genes. Although the precise molecular mechanisms have yet to be fully elucidated, it is apparent that MITF p.E318K acts as a gain of function mutation predisposing to familial melanoma. Carriers of the p.E318K variant have been identified as having a significantly higher risk of developing melanoma (ORs of 2.09–2.19), and the p.E318K mutation has been shown to cosegregate with melanoma in multiple families.54 ,55 Cosegregation was observed in some but not all family members, implying that it is a medium-penetrance melanoma variant, similar to MC1R.55 From population analysis of controls, few mutations were detected, therefore denoting MITF p.E318K as a rare population variant.55 ,57 ,58 The p.E318K mutation has also been linked with a particular phenotype, comprising non-blue eye colour, increased number of naevi and multiple primary melanomas.54 ,55 ,57 ,58
Solute carrier family 45, member 2
In contrast to MC1R and MITF, solute carrier family 45, member 2 (SLC45A2) variants are associated with darker skin colour, and appear strongly protective against melanoma. The gene product functions as a membrane-associated transporter protein, and is thought to influence pigmentation via the processing and trafficking of melanosomal proteins such as tyrosinase.59 ,60 The ancestral variant 374 L of rs16891982 has been associated with olive and dark skin, and confers a protective effect against melanoma, even for individuals with a fair phenotype (ORs of 2.37–5.50).59 ,61 ,62 This variant is more common in individuals from southern Europe and the Mediterranean region, and there is a decreasing gradient of allele frequency from the south to the north of Europe.60 ,63
Low penetrance genes
In addition to two of the known medium penetrance genes MC1R and SLC45A2, 18 other low penetrance risk loci have been associated with melanoma through GWAS (table 1).64 Agouti signalling protein (ASIP), tyrosinase (TYR), tyrosinase-related protein 1 (TYRP1) and oculocutaneous albinism type II (OCA2) are involved in pigmentation.2 ,60 ,61 ,65 ASIP encodes for an antagonist of α-MSH, which competitively binds to MC1R, thereby preventing MC1R-mediated stimulation of eumelanin synthesis (figure 5). ASIP has been variably associated with melanoma.2 Similar to MC1R RHC variants, melanoma-associated ASIP SNPs have been linked to red hair and skin freckling.66 TYR impacts eye colour and tanning response, where activity of the enzyme tyrosinase influences the ratio of eumelanin to pheomelanin, and thus TYR alterations can contribute to a fair skin phenotype (figure 5).66 TYR SNPs associated with blue eye colour and skin sun sensitivity have been significantly associated with melanoma, as have SNPs in TYRP1.2 ,66 TYRP1 stabilises the protein encoded by TYR, and therefore mutations in this gene can also affect tanning response.66 Further to known phenotypic associations with melanoma, increased risk has been reported for pigment related SNPs in the HERC2/OCA2 region on chromosome 15q13.1.67 The two SNPs most significantly associated with melanoma risk are rs1129038 and rs12913832, the latter being a key determinant of human blue-brown eye colour.67 ,68 PLA2G6 is associated with pigmentation and naevi, while CASP8, TERT, AGR3, MTAP/CDKN2A and FTO are associated with variation in naevus density.14 ,61 ,63 ,65–67 ,69–78 In addition to its role in naevus count, TERT is also associated with telomere length, as is OBFC1.73 Two loci (PARP1, ATM) are associated with DNA repair, and two others are linked to methylthiolation of tRNA and regulation of cell cycle progression (CDKAL1 and CCND1, respectively).65 ,73 ,79 ,80 Four other loci: ARNT/SETDB1, CYP1B1, MX2 and TMEM38B/RAD23B, are associated with melanoma but via uncertain mechanisms.65 ,73 ,79
Melanoma loci identified through GWAS
Genetic modifiers and interactions
Overall risk in familial melanoma is modified by the pooled contribution of many factors, including other genes, phenotypical characteristics and the environment. The addition of modifiers or interactions can influence the penetrance of a certain allele, and contribute to increased, or decreased, melanoma susceptibility.
Gene-gene
Epistasis is a gene-gene interaction, where the effect of a particular gene depends on the presence of another modifier gene. Epistasis can also be linked to multiple genes, where a certain genetic background may be essential for subsequent gene expression.81 A number of epistatic mutations likely contribute to the polygenic inheritance of melanoma.
Further to its contribution as an independent risk gene for melanoma, MC1R variants act as genetic modifiers by increasing the penetrance of CDKN2A mutations. A recent meta-analysis showed that melanoma risk doubled for patients with mutations in CDKN2A and MC1R compared with mutated CDKN2A alone, and that carriers of multiple MC1R variants were even more likely to develop melanoma.3 For potential interactions of MC1R with other genes located near CDKN2A, all 10 recently identified candidate polymorphisms on chromosome 9p21 did not show any significant association on interaction analysis.11
Telomere length has been also investigated in relation to CDKN2A status, following previous associations between cutaneous melanoma and longer telomeres. In contrast to non-carriers, the study failed to show a link between telomere length and melanoma for carriers of CDKN2A mutations, either suggesting a divergent melanoma pathway in these individuals, or more likely, insufficient power to detect an association.40
Interaction between the MITF p.E318K allele and MC1R RHC variants has been variably reported, and there does not appear to be an interaction in the majority of patients.58 ,82 However, one patient in an Australian cohort with a p.E318K allele and MC1R homozygous R/R genotype developed three amelanotic melanomas, suggesting a genetic interaction as the source of this phenotype.58 An analysis of 33 candidate polymorphisms in several pigmentation genes and the vitamin D receptor (VDR) gene identified significant epistatic effects between MC1R and TYR, and SLC45A2 and VDR, among others.12
MC1R has been proposed to also interact with somatic BRAF p.V600E mutations to drive melanomagenesis, likely by allowing cells to bypass senescence.83 In vivo studies have demonstrated that the simultaneous expression of BRAF p.V600E and MC1R depletion results in greater melanocyte growth and tumour formation compared with either factor alone.83
Gene-phenotype
Pigmentation traits with less melanin are linked to melanoma via reduced protection against UVR. Phenotypic risk factors for melanoma in the general population include blue or green eyes, fair or red hair, fair skin with increased sun sensitivity and an inability to tan, high numbers of naevi, and atypical naevi. Several genetic variants predisposing to pigment and naevus phenotypes have been identified in the general population, which in turn have been implicated in predisposition to melanoma.45
In addition to the effect on DNA repair, the variant MC1R-mediated RHC phenotype of red hair, pale skin, and an inability to tan confers melanoma susceptibility by increased potential for sunburn and UVR damage.45 The medium penetrance risk gene MITF has been associated with the phenotypic characteristic of high naevus count.55 ,58 In an Australian study, carriers displayed significantly higher counts of naevi greater than 5 mm, but without distinct dermoscopic naevus signature patterns.58 For carriers of the p.E318K mutation, there was an association with non-blue eye colour but no association with other known phenotypic characteristics, including skin colour, hair colour and freckling.55
Gene-environment
The most significant independent environmental risk factor for melanoma is UVR exposure, and a potential interaction between geographical location and CDKN2A penetrance has been observed. A large international study of families from three continents found significant variation in mutation penetrance depending on geographical location, likely correlating with associated UVR exposure.1 By age 50 years, mutation penetrance reached 0.13, 0.50 and 0.32 in Europe, the USA and Australia, respectively. By age 80 years, it was 0.58 in Europe, 0.76 in the USA and a staggering 0.91 in Australia.1 Although these penetrance rates appear to correspond with latitude and hence UVR exposure, it is possible that varying penetrance of different region specific CDKN2A variants or co-inheritance of other genetic modifiers could contribute to the differences.
Tobacco smoke has been linked to increased penetrance of CDKN2A for pancreatic, upper gastrointestinal and respiratory cancers, and it is hypothesised that it may also affect CDKN2A penetrance for melanoma.4
Familial melanoma mutations and risk of other cancers
Some familial cutaneous melanoma predisposition genes have also been linked to risk of other tumour types, where the incidence of specific cancers occurs within melanoma families at rates greater than expected by chance (table 2).
Melanoma predisposition genes and associations with other cancers
The most extensively documented association is between CDKN2A and pancreatic cancer, although associations have been noted for a range of other cancers.4 ,5 ,84 ,85 A study of carriers of the Swedish p.Arg112dup CDKN2A founder mutation found significantly increased risk of pancreatic cancer, upper digestive (oral cavity, tongue, pharynx, larynx, oesophagus, stomach, liver, gall bladder) cancers and respiratory (bronchi and lung) cancers.4 At age 80 years, 53% of carriers were reported to have at least one of these specific cancers.4 Interestingly, the risk of cancer was significantly higher in individuals who had ever smoked, compared with carriers who had never smoked.4 Upper gastrointestinal and respiratory tissues are particularly sensitive to carcinogens, and exposure to tobacco smoke and other environmental carcinogens may increase the penetrance of CDKN2A in these cancers in a similar manner to UVR and melanoma. An international study reported an increased risk of all non-melanoma cancers in first degree relatives of CDKN2A mutation carriers.84 For mutation carriers, the lifetime risk of any cancer other than melanoma was estimated at 59% by age 85 years.84
Further to CDKN2A variants and melanoma risk, the 9p21 locus has been linked to a variety of other cancers. An analysis of eight different GWAS identified several significant SNPs in this region, including some variants associated with multiple cancers.86 Of particular note may be the CDKN2A intronic rs3731239 SNP, which was associated with oesophageal squamous cell cancer, gastric cancer and breast cancer.86 Although these results to do not directly relate to specific melanoma risk SNPs, it is interesting to consider the potential impact of this region to cancer susceptibility more generally, and as a potential site for other novel cancer predisposition variants.
An association with multiple cancers has also been indicated for POT1.38 ,39 Other cancer types include breast cancer, small cell lung cancer, endometrial cancer and brain tumours, which have been observed in POT1 mutation carriers and their untested family members. There may also be a link with chronic lymphocytic leukaemia (CLL), which has somatic mutations in POT1 at relatively high frequency, the majority of which affect the OB folds, a finding that is in keeping with alterations detected in the recent melanoma studies.38 ,39 ,87 ,88 One POT1 mutation carrier had a history of melanoma and CLL, and it is possible that a variant exists that could affect a portion of the OB fold domain that is relevant to the development of melanoma and CLL.38
Additionally, POT1 has recently been implicated in the development of glioma.89 Three novel protein-changing variants have been described, each found in one family with a high case density of glioma. In one family with six carriers and one obligate carrier of p.G95C, three individuals developed glioma at young ages.89 Of six carriers in a family with a p.E450X mutation, two were affected by glioma.89
Glioma has previously been tentatively associated with melanoma following the observation of more melanoma cases than expected in glioma families.90 Although the underlying basis for susceptibility is uncertain, analysis of potential glioma susceptibility loci by GWAS has identified variants in chromosome 9p21 near CDKN2A and CDKN2B.91 ,92 The glioma candidates are not in the same linkage disequilibrium block as the CDKN2A melanoma gene, but it suggests the possibility that this region may account for shared predisposition to both cancers.
More cancers than expected have also been found in families carrying ACD and TERF2IP mutations, which like POT1, affect the shelterin complex. Although the numbers are too few to be statistically significant, additional cancers in carriers with melanoma include lung, breast, bowel and haematological malignancies, suggesting a possible ACD/TERF2IP associated spectrum of cancers.41
Somatic TERT promoter mutations have been found in a wide range of different cancer types, and the occurrence of multiple additional cancers in individuals affected by a novel germ line promoter mutation suggests that these other cancers could be due to dysregulation of TERT.42 ,43 One individual was diagnosed with endometrial cancer at age 27 years and melanoma at age 30 years. A second affected family member developed melanoma at age 20 years, then subsequently had ovarian cancer, RCC, bladder cancer, mammary carcinoma and bronchial carcinoma before her death at age 50 years.43 It has been suggested that the nucleotide sequence change in the germ line variant creates a binding motif similar to the one already used by the ternary complex factor Elk1, which has been demonstrated as a transcriptional regulator in breast, cervical and endometrial cancers.43 ,93–95 Although a tenuous link, this could help explain gender related differences as well as the presence of endometrial, ovarian and breast cancers.
A bidirectional association has been established between melanoma and RCC for sporadic cases, and a number of familial melanoma studies have also noted an over-representation of this cancer.96 RCC and pancreatic cancer have been linked to the p.E318K mutation in MITF, and a potential connection between melanoma and lymphoma has been noted.54 ,55 ,57 Mutation p.E318K upregulates hypoxia inducible factor, which has been identified as the downstream target of other known RCC predisposition genes.54 BAP1 has also recently been associated with the development of RCC. In an analysis of 60 French families with BAP1-reminiscent cancer clustering, RCC-affected individuals were identified in 6 out of 11 families with germ line BAP1 mutations.26 A novel variant has been detected in one American family with multiple cases of RCC but no other cancers, suggesting that germ line BAP1 mutations may rarely predispose solely to RCC.97
The high density of cancer in families affected by germ line BAP1 mutations suggests that this gene is a critical regulator of oncogenesis for the tumours identified.30 The numerous functional domains of the BAP1 protein present a range of potential sites for mutation. Therefore a number of germ line variants may exist, each possibly contributing to a different collection of cancers. Further to the heterogeneity of BAP1 mutations, it is likely that modifier genes and environmental factors also impact the cancer phenotype in BAP1 families. A BAP1 cancer cluster comprised of cutaneous/ocular melanoma, atypical melanocytic proliferations, and other internal neoplasms such as mesothelioma has been proposed as a particular syndrome.23 However, further studies have implicated a range of other cancers as part of a possible BAP1 spectrum. In addition to RCC, possibly associated neoplasms include lung adenocarcinoma, meningioma, paraganglioma, breast cancer, neuroendocrine tumours, gastric cancer and basal cell carcinoma.22 ,24 ,25 ,27 ,28 These findings hint at the prospect of many other BAP1 associated cancers as more families are identified.
Familial cancer syndromes and melanoma risk
A number of other rare autosomal familial cancer syndromes have been described, characterised by the occurrence of multiple cancers including melanoma. These include Li-Fraumeni syndrome, xeroderma pigmentosum, Werner syndrome and familial breast cancer. Li-Fraumeni syndrome is linked to TP53, and germ line mutations are associated with breast cancer, bone and soft tissue sarcomas, brain tumours and adrenocortical carcinomas.98 ,99 The inclusion of melanoma in the syndrome has been controversial, however a handful of melanoma cases have been reported, including one patient with a germ line TP53 mutation who presented with multiple primary cutaneous melanomas.100 In contrast, xeroderma pigmentosum is an autosomal recessive condition caused by mutations in one of eight nucleotide excision repair genes, and the DNA repair function they encode is crucial to the cellular response to UVR-induced DNA damage.101 Coupled with UVR damage, this failure in DNA repair predisposes to increased sun sensitivity and skin cancers. A 2000-fold and 10 000-fold increase in melanoma and non-melanoma skin cancers, respectively, has been reported, as well as an increase in neural system cancers.101 Werner syndrome is also autosomal recessive, and loss of function mutations in the WRN gene lead to premature aging and multiple cancer susceptibilities, with the spectrum comprising thyroid cancer, melanoma, meningioma, sarcomas and leukaemia.102 Analysis of tumours other than breast cancer in carriers of BRCA1 or BRCA2 mutations has shown that BRCA2 defects are associated with 2.6-fold and 99.4-fold increased risks of cutaneous and uveal melanoma, respectively, but there is no increase in melanoma risk associated with BRCA1 mutation.103 ,104
Clinical implications and future practice
The identification of cancer predisposition genes by genetic testing is typically only recommended when the results influence clinical decisions and treatment can be implemented to prevent or improve clinical outcomes.105 Genetic testing in melanoma is therefore controversial, due to the relatively low frequency of high penetrance mutations and the contribution of multiple additional factors that modulate melanoma risk. Despite this, heightened surveillance and more regular skin checks could be a useful outcome for a patient with a known susceptibility.
One of the main benefits encountered from genetic testing is that it may prompt useful discussions about melanoma risk, early detection and prevention with multiple family members. The impact of melanoma risk discussion on the effect on future sun safety behaviours has been demonstrated in a group of family members identified for CDKN2A testing.106 Two years following genetic testing, individuals sustained improvements in daily sun protection and fewer sunburns, with no diminution after a negative test result.106 Although this study is subjective, it highlights the potential positive impact of increasing awareness and education. Counselling may therefore form an opportunistic intervention to motivate preventative behaviours and minimise UVR exposure risk.
With time, it is anticipated that the data pool of presently known variants will expand, which will be particularly important for analysis of rare variants in a wider population. For families with a high cancer burden but no carriers of previously identified predisposition genes, next-generation sequencing will be key to identifying potential novel high penetrance variants and narrow the present knowledge gap. If future studies indicate clinical utility for genetic testing, it is likely that only high penetrance predisposition genes would be prioritised for gene panels dedicated to melanoma risk evaluation. Although the epistatic effect of MC1R variants on CDKN2A penetration has been noted, it is less likely that low penetrance risk or medium penetrance risk genes would be used as routine screening tests due to the uncertainty of predicting the clinical outcome of disease development.3
Progressing from attribution of melanoma risk, future practice in familial melanoma may involve novel susceptibility genes as a basis for development of early detection strategies. The possibility of combining clinical and genetic information for prognostic estimates has been proposed, where a novel logistic regression model of two significant SNPs, histological tumour type and stage at diagnosis had an improved discrimination of 3-year melanoma recurrence compared with histology and stage alone.107 A recently published study analysed 2339 SNPs in 14 autosomal genes of the Fanconi anaemia pathway, which is involved in the cross-link repair of DNA. Four SNPs were significantly associated with reduced overall survival and melanoma-specific survival, and combination of these factors with tumour stage and Breslow thickness further refined 5-year predictive ability.108
Although the potential for targeted treatments directed at germ line mutations seems unlikely, it may be plausible in the future, particularly for high penetrance genetic variants with germ line and sporadic manifestations.
Conclusion
Overall, the landscape of melanoma risk genes is becoming gradually less mysterious, with the addition of BAP1, POT1, ACD, TERF2IP and TERT to the known high penetrance melanoma risk genes CDKN2A and CDK4. Ongoing studies of recently identified pigmentation genes in a wider population will be highly significant in their independent risk and the additional risk conferred by gene-gene and gene-phenotype interactions. Novel candidate genes are promising, however there likely still remains a great many to be elucidated. The contribution of melanoma risk genes to other cancers is particularly important for families with observed cancer clustering, where novel genes may also predispose to other cancers. In the future, it is plausible that melanoma risk genes could be used for genetic counselling of melanoma as well as the other cancers they influence. The most important outcome of familial melanoma research will be in clinical application, and even without genetic testing, awareness of the hereditary component of melanoma is likely to improve health promotion and advocacy as part of holistic patient care. Future research will continue to validate known risk genes in wider populations, and will also aim to discover novel predisposition genes for the large percentage of families with a high case density but no identified presently known genes. Although routine genetic testing is currently not recommended due to the complex polygenic interplay that influences the clinical picture of melanoma, the potential for predisposition genes to be utilised as screening tools, for prognostic information, and as targets for treatment may be important in future practice.
Acknowledgments
NKH is supported by a fellowship from the National Health and Medical Research Council of Australia. The authors are extremely grateful to Dr Ulrikke Lei, MD, Senior Consultant in Dermatology, Herlev and Gentofte Hospital, Hellerup, Denmark for kindly providing some of the clinical photographs, and to Dr Matthew Law, QIMR Berghofer Medical Research Institute for statistical analysis of low penetrance risk loci.
References
Footnotes
Contributors Preparation of initial manuscript and figures: JR. Revision and review of all drafts: JR and NKH. Sourcing of clinical photographs: KAWW. Drafting of final versions of the manuscript and final approval of manuscript: JR, NKH and KAWW.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.