Article Text

Abstract
Screening for Fabry disease (FD) reveals a high prevalence of individuals with α-galactosidase A (GLA) genetic variants of unknown significance (GVUS). These individuals often do not express characteristic features of FD. A systematic review on FD screening studies was performed to interpret the significance of GLA gene variants and to calculate the prevalence of definite classical and uncertain cases. We searched PubMed and Embase for screening studies on FD. We collected data on screening methods, clinical, biochemical and genetic assessments. The pooled prevalence of identified subjects and those with a definite diagnosis of classical FD were calculated. As criteria for a definite diagnosis, we used the presence of a GLA variant, absent or near-absent leukocyte enzyme activity and characteristic features of FD. Fifty-one studies were selected, 45 in high-risk and 6 in newborn populations. The most often used screening method was an enzyme activity assay. Cut-off values comprised 10–55% of the mean reference value for men and up to 80% for women. Prevalence of GLA variants in newborns was 0.04%. In high-risk populations the overall prevalence of individuals with GLA variants was 0.62%, while the prevalence of a definite diagnosis of FD was 0.12%. The majority of identified individuals in high-risk and newborn populations harbour GVUS or neutral variants in the GLA gene. To determine the pathogenicity of a GVUS in an individual, improved diagnostic criteria are needed. We propose a diagnostic algorithm to approach the individual with an uncertain diagnosis.
- Fabry disease
- Mass Screening
- Neonatal screening
- Genetic testing
- Prevalence
Statistics from Altmetric.com
Introduction
Fabry disease (FD) (McKusick 301500) is an X-linked lysosomal storage disorder with an estimated birth prevalence of 1:40 000–170 000,1–3 based on clinical ascertainment. The cause of the disorder is a deficiency of the hydrolase α-galactosidase A (AGAL-A), resulting primarily in storage of globotriaosylceramide (CTH, GL-3 or Gb3).1 More than 600 variants in the GLA gene have been described.4 ,5 Most of these appear in single families. In the hemizygous male, the disease may present during childhood or adolescence with characteristic features, such as acroparesthaesia, anhidrosis, disseminated angiokeratoma, cornea verticillata, gastrointestinal symptoms and (micro)albuminuria.1 At a later age, progressive renal failure, hypertrophic cardiomyopathy and cerebrovascular disease (eg, stroke) can occur.1 Heterozygous women may also be affected, yet they generally demonstrate a more variable and attenuated phenotype. In men and women, life expectancy is diminished, although it is more apparent in men.6 ,7
An increasing number of screening studies in high-risk populations as well as newborn, screening studies have been performed since enzyme replacement therapy (ERT) became available; currently, ERT is the only approved causative treatment. These studies revealed an unexpected high number of individuals with mutations or genetic variants of unknown significance (GVUS) in the GLA gene and/or decreased AGAL-A activity. For example, in newborn screening studies, birth prevalences up to 1:1250 were reported, about 30 times higher than previously estimated.8–11 While studies differed in methodology, it appeared that most male patients with such GLA variants demonstrated residual enzyme activity, in contrast to the absent or near-absent enzyme activity in classically affected males.
To establish a definite FD diagnosis in a classically affected male FD patient is usually straightforward. Such patients often have multiple characteristic FD signs and symptoms, very low or completely absent AGAL-A activity, increased Gb31 and lysoglobotriaosylceramide (lysoGb3) concentrations in plasma and urine12–17 and a pathogenic mutation. However, many of the individuals detected in high-risk screening studies express only a single non-specific symptom, such as left ventricular hypertrophy or stroke, the symptom that was used as the entry criterion for screening. This symptom could be completely unrelated to the detected GLA variant or could be caused by a non-classical form of FD. A patient with an early diagnosis and a classical FD phenotype might benefit from timely treatment and counselling. Equally important, the individual with a GLA variant, who does not have FD, should be identified to avoid unnecessary treatment. It is also important to note that, in contrast to classical FD, in non-classically affected patients with FD, the natural history and effectiveness of ERT are unknown.
Thus, there is an urgent need to gain insight into the frequency of definite classical Fabry cases detected by screening studies and the prevalence of those with a GVUS. Individuals with a GVUS may turn out to have non-classical FD or no FD at all.
Therefore, we performed a systematic review on Fabry screenings studies in newborns and high-risk populations. We aimed to systematically interpret the significance of the GLA gene variants and/or the deficiency of the AGAL-A enzyme activity in identified individuals and their family members, and to estimate the prevalence of definite classical cases and cases with a GVUS according to our proposed criteria for a definite FD diagnosis in this article. A strategy to optimise the diagnosis of FD is proposed in individuals with a GVUS in the GLA gene.
Methods
Search
We searched PubMed and Embase until 27 November 2012, using the search terms ‘Fabry disease’ AND ‘Screening’ OR ‘Prevalence’. The search was extended with synonyms for FD and Mesh terms or Headings.
We selected full-text articles in peer reviewed journals in all languages that aimed to identify individuals with FD in a well-defined cohort (eg, stroke, left ventricular hypertrophy, renal disease, newborns) and with clear screening methods. References were cross-checked for additional relevant papers. Papers with single cases or family studies were excluded.
Data collection
All reported alterations in the coding sequence of the GLA gene and intronic variants are referred to as genetic variants. When the pathogenicity of the variant is not known, it is referred to as GVUS.
Data on inclusion and exclusion criteria for screening and laboratory data were collected. Biochemical analyses for diagnostic purposes included AGAL-A enzyme activity in leucocytes, plasma and serum. In all cases, the enzyme assay for AGAL-A as described by Mayes et al18 was used. In screening studies, the source of enzyme was either plasma, serum or dried blood spots (DBS). In confirmatory studies, mostly leukocytes were used. While a deficiency of AGAL-A can be reliably established with all enzyme sources, leukocytes are clearly superior to estimate residual AGAL-A activities. Therefore, only leukocyte data were used in our study for estimating the residual activities. Nevertheless, caution should be taken when this assay is used to estimate the true residual activity in vivo since the contribution of AGAL-B may not always be completely suppressed in the assay and the artificial substrate has different properties than the natural substrate. Reference values for healthy controls and cut-off values for positively screened individuals were collected. Additionally, the results of molecular analysis of the GLA gene were recorded. We registered the total number of individuals in the screening study, the number of positively screened individuals in the first screening test, and the number of true and false positives after the confirmatory test. All data were specified for males and females if available. For all true positives the following characteristics were collected: gender, age, GLA genotype, residual GLA enzyme activity (plasma, leukocyte and serum), lysoGb3 and the presence of phenotypic FD characteristics angiokeratoma, acroparesthaesia and/or cornea verticillata. Anhidrosis was not used as a characteristic because of the subjective nature of the symptom. If the author performed a pedigree analysis, we collected data on the presence of these characteristics in family members.
In order to recalculate the prevalence of FD, we systematically classified all individuals with a GLA variant (true positives) into three groups based upon the criteria in table 1 being: ‘Classical’, that is, individuals with a definite diagnosis of classical FD, ‘Uncertain/Neutral’, that is, individuals with a GVUS or an as-yet unidentified neutral variant, and ‘Unknown’ if insufficient data were available (no reported leukocyte AGAL-A enzyme activity and no information on characteristic symptoms of the individual). Individuals with the GLA variants p.E66Q and p.D313Y were classified as ‘Uncertain/Neutral’. Based upon extensive laboratory and clinical studies, these GLA variants are considered polymorphisms, that is, neutral variants.19–21 Other GLA gene variants have been characterised in the literature as possible neutral variants, for example, p.A143 T.18 For other variants, evidence for pathogenicity is limited, for example, p.R112H.19 lysoGb3 or Gb3 in plasma or urine were not used in the criteria because very few studies report results on these biomarkers.
Criteria for classification of screen positive individuals
Monserrat et al22 described three males with the p.D313Y variant, who were not included in the estimate of the prevalence by the author. For consistency, we included these patients in our calculations, and classified them as ‘Uncertain/Neutral’. Porsch et al23 identified two individuals with a low DBS AGAL-A enzyme activity and confirmed a diagnosis in one (c.30delG, personal communication). In the second male identified by Utsumi et al, the authors did not find a variant in the GLA gene in one male, but this individual was included in our analysis because a low AGAL-A activity of 16% of the normal mean in the daughter suggested a pathogenic mutation.24 Rolfs et al reported that 28 individuals had a ‘biologically significant mutation’, but did not disclose further information.25 These individuals were included and categorised as ‘Unknown’ in the recalculation.
Data on financial support were collected from the publications.
Statistical analysis
Studies in high-risk populations were categorised according to the entry criterion for screening, that is, hypertrophic cardiomyopathies (HCM, a cardiac disease), end-stage renal disease (ESRD, kidney disease including renal transplantation), stroke/small fibre neuropathy (SFN) (neurological disease, not including migraine) or other (migraine and atherosclerosis).
Pooled prevalence of individuals with a mutation or variant in the GLA gene was calculated. A recalculation was performed for all individuals who were categorised as ‘Classical’. All calculations were specified for gender. Studies in which gender was not specified, were excluded in the gender specific prevalence calculation, but were included in the total prevalence calculation.
Exome variant server
All genetic variants were searched for in the exome variant server (EVS),26 and the allele prevalence was reported for the most frequent variants.
Results
Literature search and selection of papers
The search identified 538 papers, of which 57 were selected based on title and abstract. Cross-checking references revealed two more relevant papers.27 ,28 Articles in which only ethical issues regarding screening, or screening methods were discussed, reviews or papers not including FD, were excluded. Eight papers were excluded because they did not fit the criterion of screening a well-defined population (eg, left ventricular hypertrophy, stroke, renal insufficiency, newborns),29–32 because only patients with a high suspicion of FD within a high-risk population were selected,33 because an insufficiently defined screening method was used,34 ,35 or because the study was performed in the same study centre36 as another screening study37 and overlap in the studied population could not be excluded.
Patient characteristics and screening methods of included papers
The HCM studies (n=12)22 ,27 ,28 ,38–46 included subjects with left ventricle hypertrophy according to local criteria. The ESRD studies (n=22)23 ,24 ,37 ,47–65 concerned individuals on haemodialysis, continuous peritoneal dialysis, kidney transplantation and/or chronic kidney disease. Stroke studies (n=9)25 ,66–73 included patients with (first or cryptogenic) stroke, transient ischaemic attack, white matter lesions and/or basilar dolichoectasia or, in one study, SFN. Two studies that were labelled as ‘other’ were performed in persons with migraine, and premature atherosclerotic males, respectively.74 ,75 Six studies on newborn screening were included.8–11 ,76 ,77
Table 2 summarises data on populations and screening methods. A DBS enzyme activity assay was used as the first screening method in the majority of the included papers. The cut-off value ranged from 20% to 55% of the mean reference value in men and from 30% to 80% in women, where specified.
Population and screening method
In most studies criteria for a definitive diagnosis for FD were not specified.
Financial support and publication rate
Twenty-three (45%) studies were financially supported by companies that market treatments for FD: Shire HGT, formerly TKT, (8/51, 15%) or Genzyme, a Sanofi company, (15/51, 29%). One study was sponsored by both companies (2%). Thirteen (25%) studies were supported financially by (personal) grants or other sources. In 14 (27%) studies, no information on financial support was provided.
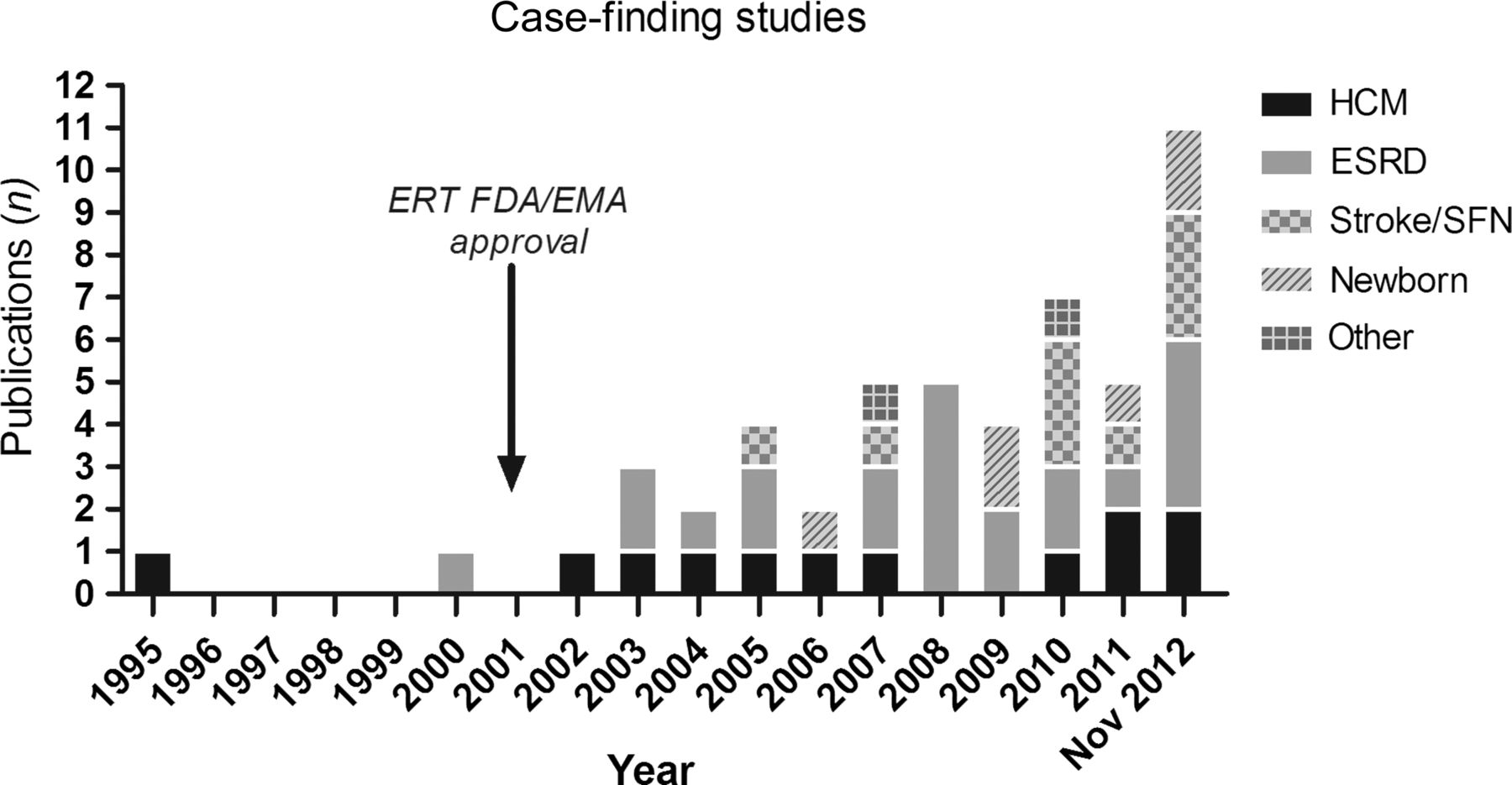
The number of published papers per year increased steeply after ERT became available in 2001 (see figure 1).

Screening studies per year and category. EMA, European Medicines Agency; ERT, enzyme replacement therapy, FDA, US Food and Drug Administration.
FD prevalence in newborns and GLA variants
The pooled prevalence of individuals with a variant in the GLA gene in newborns was 0.04% (number screened n=397 271, GLA variant n=153) while the prevalence in individual studies ranged from 0% to 0.04%. A recalculation of ‘Classical’ FD could not be performed because the newborns expressed no characteristic FD signs, and for the majority no family data were available. Data on lysoGb3 were not available.
In the Taiwanese newborn screening studies,9 ,10 the intronic splice site variant IVS4 + 919G >A was found in 83% (n=100/120) of newborns with a GLA variant. In European newborn studies, the most frequent genetic variants were: p.A143 T (n=10/29), p.R112H and p.D313Y (both n=2/29). A complete list of the genetic variants is presented in the online supplementary material.
FD prevalence in high-risk populations and GLA variants
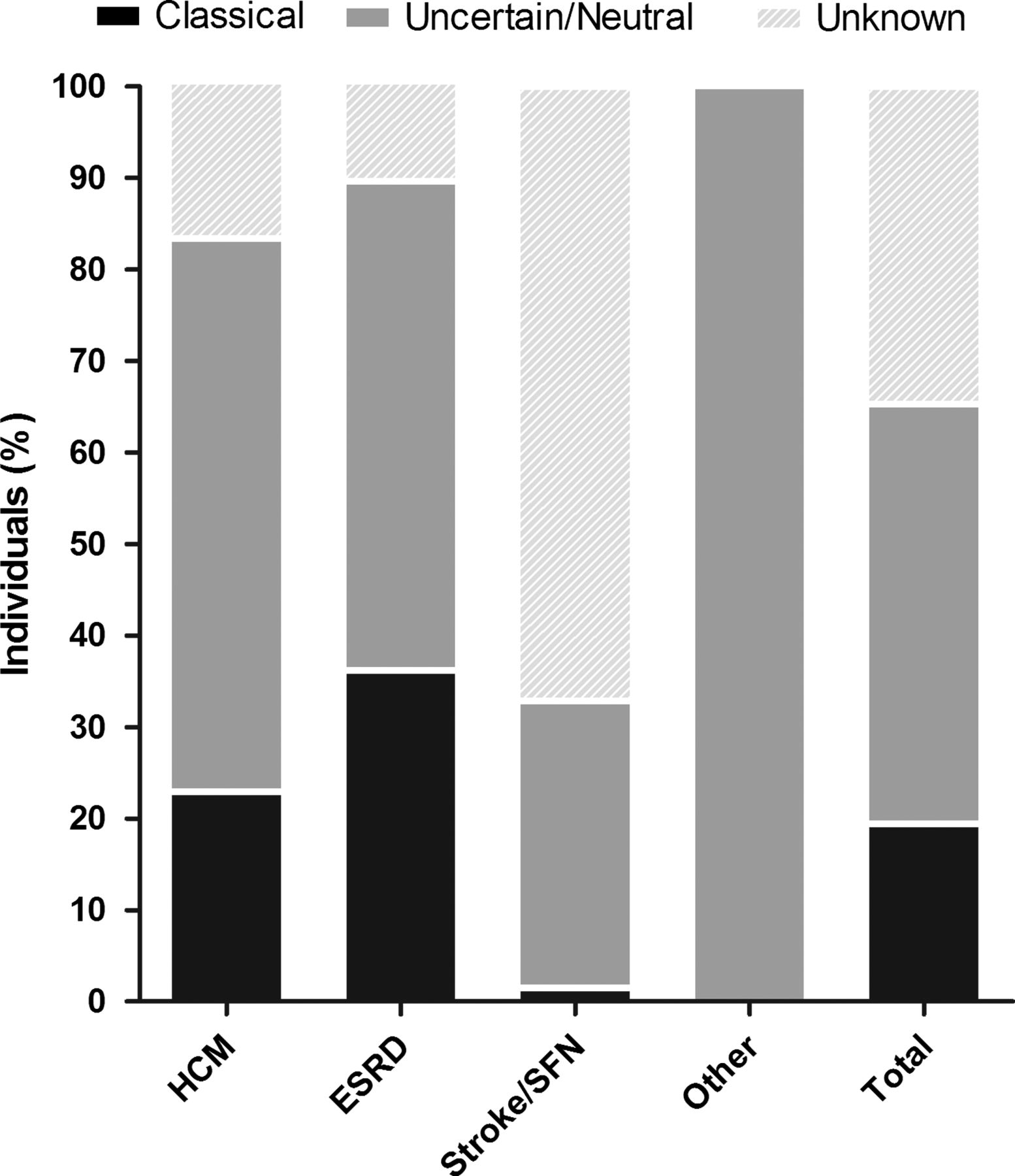
Totally, 174 out of 28165 screened individuals were found to have a GLA variant, revealing an overall prevalence of 0.62% (individual study results ranged from 0% to 12%). Of these, 33 individuals met the criteria of ‘Classical’ FD, resulting in a prevalence of 0.12% (n=33/28165). Most individuals who were identified were categorised as ‘Uncertain/Neutral’ 0.30% (n=84/28165) or ‘Unknown’ 0.20% (n=57/28165). In the uncertain/neutral group, 42 individuals harbour the neutral variants p.E66Q or p.D313Y, and 42 harbour a GVUS, accounting for a pooled prevalence of GVUS of 0.15% in the screened population. Details of the prevalence calculation are shown in table 3 and figure 2.
Prevalence of individuals with a GLA variant and ‘Classical’ Fabry disease (FD)
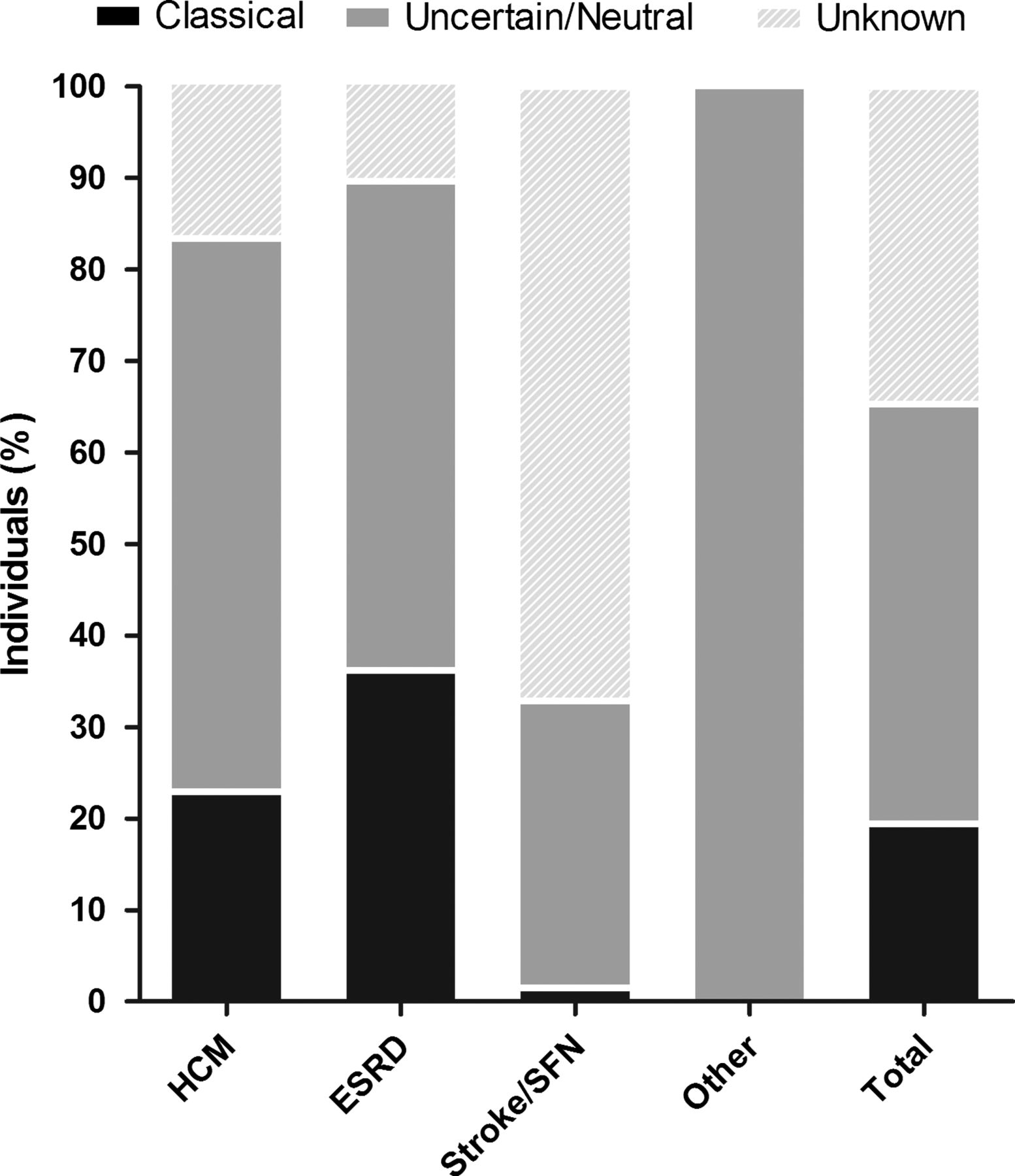
Distribution of individuals with a variant in the GLA gene after classification.
Data on lysoGb3 were available for five individuals. Dubuc et al69 reported an initial slightly elevated lysoGb3 in plasma (1.97 ng/mL, reference <1.84) but a normal finding on second analysis in one individual. This male individual had a combination of splice site variants in the GLA gene (categorised as ‘Unknown’). LysoGb3 was not available for the other six individuals in that study. Four female individuals identified by Tanislav et al72 also harboured a combination of intronic variants in the GLA gene and had a reported increased lysoGb3 in plasma (0.59–1.45 ng/mL, reference mean 0.47, SD 0.06).
Frequently encountered variants in the GLA gene were: p.D313Y (n=24, 14%) with an allele frequency in the EVS (EVS, in a total of 10 563 alleles)26 of 0.3% , p.E66Q (n=18, 11%, not present in EVS), p.A143 T (n=16, 9%, allele frequency in EVS 0.09%), p.R118C (n=11, 6%, allele frequency in EVS 0.04%), p.N215S (n=8, 5%, not present in EVS), p.M296I (n=6, 4%, not present in EVS) and p.R112H (n=3, 2%, allele frequency in EVS 0.01%). A complete list of the genetic variants and characteristics is presented in the online supplementary material.
Discussion
Screening for FD in high-risk groups reveals an overall prevalence of subjects with a variant in the GLA gene of 0.6%. In newborns, the prevalence is 0.04%. The current study demonstrates that a significant number of these individuals harbour GLA variants of unknown significance. As the number of screening studies has increased tremendously since 2001, the number of patients with these GLA variants of unknown significance also rises accordingly. Many studies (45%) were financially supported by industries that market ERT. This may have contributed to the increasing number of studies that screened for FD after ERT became commercially available.
Prevalence in newborns
Newborn screening studies in Europe and in Taiwan revealed a significantly higher prevalence of FD (as high as 1 in 1250) than was expected based on previous calculations.8–11 ,76 ,77 In two Taiwanese studies, 100/120 (83%) identified individuals showed an intronic variance of uncertain significance, IVS4+919G>A. Hwu et al demonstrated 3.6–28.5% residual leukocyte enzyme activity in men.10 Pedigree analysis revealed that in some cases, grandfathers of identified newborns who also harbour this variant show symptoms of left ventricular hypertrophy that could be associated with FD.9 This has led to the conclusion that this genetic variant may lead to a later onset or cardiac variant. However, additional data on characteristic storage in myocardial biopsies are necessary before this variant can be considered as a form of FD and poses a (limited) risk factor for cardiovascular disease.
In European and Asian newborn screening studies, several missense mutations were identified, including the frequently reported p.A143 T variant. These studies demonstrated high residual enzyme activity in expression studies, and pedigree analysis revealed non-classical disease that may or may not be caused by FD. Based upon these studies, it is likely that an estimated 90% of identified individuals in newborn studies harbour a genetic variant of unknown significance (see also the discussion below on GVUS).
Prevalence in high-risk populations
A high number of individuals with a GLA gene variant did not demonstrate characteristic features of FD. The calculated prevalence of individuals with ‘Classical’ FD in these high-risk populations was only 0.12%, or 1/1000 screened individuals. In other words, according to these criteria, for every individual with a certain diagnosis of classical FD, an estimated five individuals are identified with a GLA variant of uncertain significance or a neutral variant.
There are several limitations that may have influenced our findings. Our criteria to calculate the prevalence of individuals with a certain diagnosis of classical FD have not (yet) been validated. We deemed individuals as classically affected if a minimum of one characteristic feature was present (ie, angiokeratoma, cornea verticillata, acroparesthaesia), if AGAL-A enzyme activity was below 5%, or if there was an affected family member that showed one of the above characteristic features. However, these features can be hard to ascertain. The differentiation between atypical pains in limbs and Fabry-like neuropathic pain is difficult. Similarly, to discern angiokeratoma from senile angioma is difficult as well. We relied on the reported diagnostic accuracy of these signs or symptoms, but it is possible that this assumption has caused overestimation of the prevalence of ‘Classical’ FD. Also, an underestimation may be present if characteristic features were missed. A substantial number (38%) of individuals could not be categorised as ‘Classical’ or ‘Uncertain/Neutral’, because characteristic features and/or leukocyte AGAL-A enzyme activity were not reported. In the case of Rolfs et al, none of the 28 individuals could be categorised because of lack of data.25 LysoGb3 in plasma was available in a small subset of individuals with a combination of intronic variants in the GLA gene, and was slightly elevated. The diagnostic value and clinical relevance of such a mild elevation is yet uncertain. The lysoGb3 levels are much lower than levels that are found in classical male FD patients.17 Thus, slight elevations cannot label these patients as ‘definite classical FD’. Despite these shortcomings, we believe that these calculations give a realistic estimate of the proportions of ‘Classical’ and ‘Uncertain/Neutral’ cases that are found by screening.
Genetic variants of unknown significance
The reason to screen high-risk populations for FD is to identify patients who may benefit from adequate management, including ERT. However, in oligo-symptomatic individuals, the presence of a decreased AGAL-A activity and a GLA gene variant that is often a GVUS, need not necessarily be the cause of that symptom, especially in the absence of a family history associated with FD. Difficult clinical dilemmas have emerged as these individuals may be misdiagnosed as having FD and treated with costly ERT. The p.A143 T and p.R112H variants serve as an example. The p.A143 T variant has previously been reported to be associated with a renal variant of FD, despite a residual enzyme activity of 36% when expressed in COS cells.8 The variant has also been identified in individuals with a left ventricular hypertrophy. Terryn et al investigated 12 individuals with the p.A143 T variant including tissue biopsies and reported no storage in biopsies with light microscopy. Unfortunately, electron microscopy was not performed.78 They concluded that the variant is most likely non-pathogenic. An earlier case study also reported that Gb3 was absent in a kidney biopsy in an individual with this variant.79 Additionally, individuals with the p.A143 T variant have no increase in plasma Gb3 and lysoGb3.15 Furthermore, the AGAL-A enzyme was localised within the lysosome, suggesting normal trafficking.5 Based on these studies and the absence of characteristic symptoms of FD, we consider this GLA variant as a GVUS. However, in a single case, an individual was classified as ‘Classical’ because angiokeratoma were reported.40
The p.R112H variant has also been associated with a variable, non-classical phenotype, and individuals with this variant do not show clear elevation of lysoGb3 as seen in patients with classical FD.80 Pathological evidence of storage in podocytes has been shown by us in a subject with the p.R112H variant and renal disease (Smid et al, in preparation), but it is unclear whether this GLA variant can always explain clinical symptoms. In this context, it is important to realise that assessments of effectiveness of ERT from registry reports is also hampered by inclusion of these non-classical phenotypes or misdiagnosed subjects. For example, it should be noted that 15/20 patients in the Fabry Outcome Survey (Shire HGT) and 3/14 individuals in the Fabry Registry (Genzyme, a Sanofi company) with p.A143 T are treated with ERT.78
While for these mutations the pathogenicity is still debated, other GLA variants, such as the p.D313Y and p.E66Q, are now considered to be non-pathogenic based on in vitro expression, lysoGb3 and Gb3 concentrations in plasma, prevalence in healthy alleles, a critical appraisal of the phenotype and assessment of biopsied organs.19–21 ,81 Despite this evidence, these GLA variants are still considered pathogenic by some82 and are often included in prevalence calculations of FD in high-risk populations. These examples emphasise the need for in-depth investigations including levels of plasma lysoGb3.12 ,13 ,15–17 These studies show that the levels of lysoGb3 are strongly elevated in classically affected males, and the absence of a substantial elevation of lysGb3 should alert clinicians to perform further investigations to avoid misdiagnosis.
Proposed new diagnostic criteria for a FD diagnosis
To determine the pathogenic nature of a variant in the GLA gene in a certain individual, clear and practical diagnostic criteria are needed. In vitro expression may be helpful, but to determine the effect of a (private) genetic variant in vivo, detailed clinical assessments are mandatory. In the reviewed studies, the characteristic FD symptoms were not always clearly described and may therefore be non-specific. Characteristic features that support the diagnosis of FD should be assessed by a physician with expertise in FD. Cornea verticillata is a highly specific symptom, but concomitant medication at any time in the medical history can induce cornea verticillata, such as chloroquine and amiodarone. Also more commonly used medications, such as non-steroidal anti-inflammatory drugs, should be considered as a possible cause of cornea verticillata.83 Angiokeratoma have a clustered pattern including typical localisations, such as the umbilical and genital areas, and ought to be distinguished from simple angioma. Acroparesthaesia due to SFN can be assessed using criteria described by Biegstraaten et al84 in order to exclude non-specific pains. While anhidrosis is a specific feature, it cannot easily be objectified and should thus not be used as a diagnostic criterion for FD. Following this line of argument, a male with a characteristic pattern of symptoms, with severely decreased or absent AGAL-A enzyme activity in leukocytes, is easily identified as having a certain diagnosis of classical FD. For females, either one or more of these symptoms should be present, or an affected family member with characteristic features of FD should be identified. Plasma lysoGb3 is correlated to disease severity and has shown to be important to identify classically affected individuals.12 ,16 ,17 While this hallmark of FD is not frequently reported yet in diagnostic studies, it is highly recommended to aid in the diagnosis for both men and women.
The following criteria are proposed to identify individuals with a definite diagnosis of classical FD (see table 4). These criteria apply to all symptomatic individuals with a genetic variant in the GLA gene, and are designed to only detect individuals in whom there is no doubt about the diagnosis of classical FD.
Proposed criteria for a definite, classical diagnosis of Fabry disease (FD)
In an individual with symptoms that could be attributed to FD and harbouring any variant in the GLA gene, a male should demonstrate low (<5% of the mean reference value) AGAL-A enzyme activity in leukocytes, and have either one or both of the following criteria: a significant increase in plasma17 lysoGb3 or Gb3 in the range of classical males, or one of the described characteristic features of FD, or a family member who is identified as having a definite diagnosis of FD; a female should have either ≥1 of the following criteria: an increase in plasma lysoGb3 or Gb3 in the range of classical males, a characteristic feature of FD, or a family member who is identified as having a definite diagnosis of classical FD.
The disease manifestations in women are often heterogeneous within families with FD. This may be related to random-X inactivation; however, the literature is contradictory on this matter.85–88 A woman with a genetic variant and a (male) family member with a confirmed FD diagnosis will not necessarily develop signs and symptoms of FD. Therefore, caution should be taken in applying the proposed criteria to women, and each woman should be assessed individually.
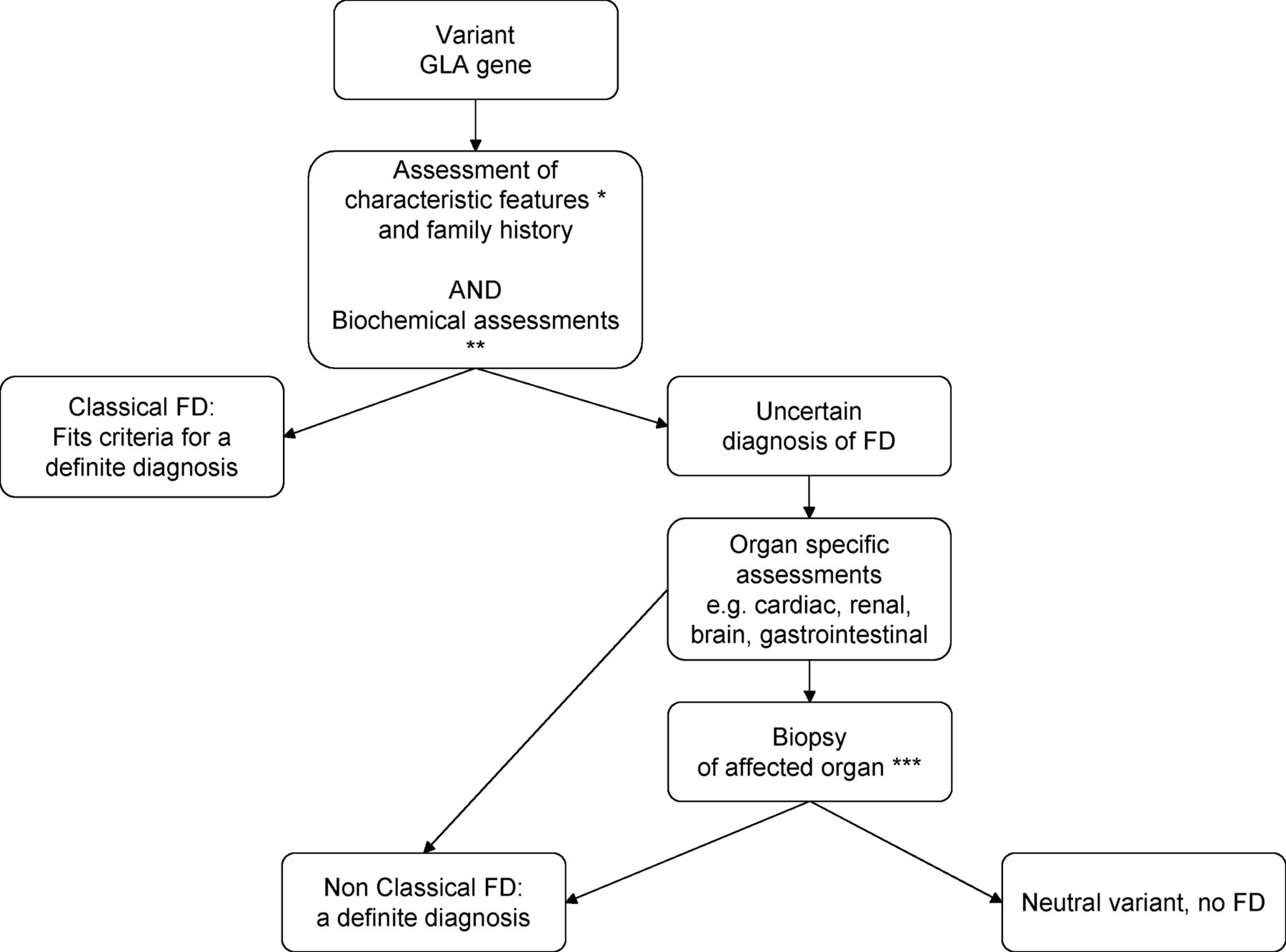
If the individual does not fit the above criteria, there is an uncertain diagnosis of FD. In such a case, we propose to follow a diagnostic algorithm (see figure 3) to ascertain if this individual has FD, that is, if the clinical symptoms that led to screening for FD are actually caused by the deficient AGAL-A enzyme. The algorithm should include biochemical analyses, functional and imaging assessments of the affected organ to look for hallmarks of FD, and where uncertainty persists, a biopsy of the affected organ to demonstrate Gb3 accumulation on electron microscopy. Other features may also be of additional diagnostic value, such as SFN as shown by quantitative sensory testing and/or a decreased intraepidermal nerve fibre count. Ultimately, such an individual may be diagnosed with FD in a non-classical form. Currently, a study is underway to determine the sensitivity and specificity of clinical, biochemical and histological assessments for each affected organ in order to delineate diagnostic algorithms that can be of use for clinicians who encounter patients with a variant in the GLA gene (Dutch trial register (http://www.trialregister.nl) NTR3840 and NTR3841).

{kind=link}
{kind=link}
{kind=link}
General diagnostic algorithm. *Ophthalmological examination (cornea verticillata), angiokeratoma, acroparesthaesia. **Leukocyte AGAL enzyme activity, plasma lysoGb3, plasma and urinary Gb3. ***For example, endomyocardial biopsy, kidney biopsy.
In conclusion, screening for FD in high-risk and newborn individuals yields many individuals with a mutation or variant in the GLA gene of unknown significance (GVUS). For these individuals, clear diagnostic criteria are needed. We strongly advise caution in screening large cohorts for FD until a clear diagnostic strategy is available so that unjustified diagnoses are avoided. In our opinion, in the absence of convincing evidence for pathogenicity in individuals with GVUS in the GLA gene, ERT should not be started. The natural history of individuals with non-classical FD is yet unknown, and the efficacy of ERT in these individuals has not been studied.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online table
Footnotes
-
Contributors All authors took part in writing and correcting the manuscript.
-
Funding This study was performed within the framework of the Dutch Top Institute Pharma (TIPharma, project number T6–504: ‘Fabry or not Fabry: valorisation of clinical and laboratory tools for improved diagnosis of Fabry disease’). TIPharma is a not-for-profit organisation that catalyses research by founding partnerships between academia and industry. Partners: Genzyme, a Sanofi company; Academic Medical Center, University of Amsterdam; Subsidising Party: Shire HGT. http://www.tipharma.com/pharmaceutical-research-projects/drug-discovery-development-and-utilisation/hamlet-study.html. The industry partners had no role in the content of this manuscript.
-
Competing interests L van der Tol has received travel support and reimbursement of expenses from Actelion, Shire HGT and Genzyme. BE Smid has received travel support from Shire HGT and Genzyme. M Biegstraaten has received reimbursement of expenses and honoraria for lectures from Genzyme, Actelion and Shire HGT. All honoraria are donated to research funds at the AMC. GE Linthorst has received reimbursement of expenses and honoraria for lectures on the management of Fabry disease from Genzyme, Actelion and Shire HGT. All fees are donated to the Gaucher Stichting, a foundation that supports research in the field of lysosomal storage disorders. CEM Hollak has received honoraria for consultancies and speakers fees from Actelion, Genzyme, Shire HGT and Protalix. All fees are donated to the Gaucher Stichting or the AMC Medical Research BV for research support. RH Lekanne Deprez and BJHM Poorthuis report no disclosures.
-
Provenance and peer review Not commissioned; externally peer reviewed.