Article Text

Abstract
Objective To describe the morphology of the pulmonary arteries in patients with pulmonary atresia, ventricular septal defect, and major aortopulmonary collateral arteries with and without monosomy 22q11.
Design A retrospective analysis of all patients with this congenital heart defect who are being followed at the University Children’s Hospital Erlangen.
Setting A tertiary referral centre for paediatric cardiology and paediatric cardiac surgery.
Patients 21 patients with pulmonary atresia, ventricular septal defect, and major aortopulmonary collateral arteries. Monosomy 22q11 was diagnosed by fluorescent in situ hybridisation using the D22S75 probe (Oncor). The morphology of the pulmonary arteries was assessed on the basis of selective angiograms.
Results 10 patients (48%) were shown to have a microdeletion in 22q11 (group I). There was no difference with respect to the presence of confluent central pulmonary arteries between these patients (80%) and the remaining 11 patients (group II) without monosomy 22q11 (91%). Patients of group I, however, more often had arborisation anomalies of the pulmonary vascular bed (90% in group Iv 27% in group II). Because of the more severe abnormalities of the pulmonary arteries, a biventricular repair had not been possible in any of the children with monosomy 22q11, though repair had been carried out in 64% of the children in group II.
Conclusions The developmental disturbance caused by monosomy 22q11 seems to impair the connection of the peripheral pulmonary artery segments to the central pulmonary arteries in patients with pulmonary atresia, ventricular septal defect, and major aortopulmonary collateral arteries, resulting in a lower probability of biventricular repair.
- catch 22
- pulmonary atresia and ventricular septal defect
- major aortopulmonary collateral arteries
- conotruncal anomaly face syndrome
Statistics from Altmetric.com
- catch 22
- pulmonary atresia and ventricular septal defect
- major aortopulmonary collateral arteries
- conotruncal anomaly face syndrome
Pulmonary atresia with ventricular septal defect represents the extreme form of tetralogy of Fallot. It may occur as an isolated lesion or as part of a genetic syndrome.1 Recent studies have shown that tetralogy of Fallot with pulmonary atresia belongs to a spectrum of conotruncal cardiac malformations that are often associated with monosomy 22q11.2 ,3 The clinical presentation of monosomy 22q11 includes patients with conotruncal anomaly face syndrome, velo-cardio-facial syndrome, and DiGeorge syndrome.4-7 More recently these syndromes have been incorporated as a group under the acronym catch 22 (cardiac defect, abnormal face, thymic hypoplasia, cleft palate, hypocalcaemia, and microdeletion 22q11).7 ,8 In the majority of cases with tetralogy of Fallot and pulmonary atresia, collateral lung perfusion is provided either by a unilateral or bilateral arterial duct or by major aortopulmonary collateral arteries.9 ,10According to the findings of Momma et al the presence of major aortopulmonary collateral arteries is associated in a high percentage with monosomy 22q11.2 Among their cohort of 49 patients with pulmonary atresia and ventricular septal defect, monosomy 22q11 was present in 21 of 34 children with collateral lung perfusion by major aortopulmonary collateral arteries.2 The purpose of our study was to determine additional anomalies of the pulmonary vascular bed and to define their impact on the treatment in the subgroup of patients with pulmonary atresia, ventricular septal defect, and major aortopulmonary collateral arteries.
Methods
All 21 patients with pulmonary atresia, ventricular septal defect, and major aortopulmonary collateral arteries who are at present being followed at our institution were included in this retrospective study. These patients were not strictly consecutive, since several children who had been diagnosed during recent years had been lost to follow up or had died.
Metaphase chromosomes of peripheral blood lymphocytes were analysed after GTG banding and by fluorescent in situ hybridisation (FISH) using the D22S75 probe (Oncor). Ten to twenty metaphase spreads were analysed in each patient. Conotruncal anomaly facies was defined as narrow palpebral fissures, broad nasal root, short philtrum, small mouth, and deformed ears, frequently associated with a hypernasal voice (fig1).2-4

Conotruncal anomaly facies in patient 3.
Information on cardiovascular anomalies in each patient was based on cardiac catheterisation with selective angiography of all major aortopulmonary collateral arteries. In those patients with central pulmonary arteries we measured the diameter of the right and left pulmonary artery just proximal to the origin of the first branch, and the diameter of the descending aorta at the level of the diaphragm. The McGoon ratio was calculated by dividing the sum of the diameters of both pulmonary arteries by the diameter of the descending aorta.
In accordance with Momma et al 2 we used the following definitions. Pulmonary artery confluence was defined as confluent right and left pulmonary arteries connected at the central portion.11 Major aortopulmonary collateral arteries were defined as vessels originating from the descending aorta or other systemic arteries connecting to central or peripheral pulmonary arteries. Patients with an arterial duct originating from the inner curvature of the distal aortic arch or from the base of the innominate artery (opposite to the aortic arch) and connecting either with the pulmonary artery confluence or with an ipsilateral central pulmonary artery were not included in the study. A high aortic arch was defined as an aortic arch reaching the third posterior rib and the clavicle on frontal cineangiograms. According to Macartneyet al 12 ,13 we defined the pulmonary blood supply as unifocal when all pulmonary segments were connected to confluent central pulmonary arteries, representing a common pathway for all blood flow to the lungs, or asmultifocal when there were segments not connected to the central pulmonary arteries and which were perfused exclusively by major aortopulmonary collateral arteries. The arborisation of each pulmonary artery was described in terms of its connection to the pulmonary artery segments.10 Normal or complete arborisation consisted of the central or proximal extrapericardial portions of each pulmonary artery being connected distally to 10 pulmonary artery segments.10
Statistical differences were evaluated using the Fisher’s exact test.
Results
CYTOGENETICS
Chromosomal analysis after GTG banding was normal in 20 patients. One patient was noted to have a fragile site on chromosome 16 (46,XY,fra(16)(q22.1)) which was present in his asymptomatic mother as well. In 10 patients (group I) FISH revealed a microdeletion (fig 2) in 22q11 (hemizygosity). All these patients had conotruncal anomaly face syndrome (table 1). The mean age of these patients was 9.7 years, while the mean age of the remaining 11 patients who were negative for the deletion (group II) was 9.8 years. One of the patients who were negative for the deletion had bilateral choanal atresia but none had clinical signs of conotruncal anomaly face syndrome. Chromosomal analysis and FISH were available in both parents of four, and in the mothers only of a further three, children with microdeletion. In three patients the microdeletion at 22q11 was present in the mother as well. These three mothers had conotruncal anomaly facies with mild learning deficiency. One of our patients with a microdeletion at 22q11 had a monozygotic twin brother (concordant for the microdeletion) who presented with classical tetralogy of Fallot. Both parents of this patient had normal chromosomes.

Metaphase spread after FISH with D22S75 probe in patient 5. The normal chromosome 22 has four fluorescein signals (arrow) representing the control (D22S39) and catch 22 regions (D22S75) on both chromatids. The chromosome 22 with a deletion has only two signals (open arrow) representing the control probe, but absent signals at 22q11.
Patient data
ANGIOGRAPHIC FINDINGS
One patient in group I also had mitral stenosis associated with an atrial septal defect (table 1). A persistent left superior vena cava draining to the coronary sinus was present in three patients in group II. One further patient in this group had partial anomalous pulmonary venous drainage of the right upper pulmonary vein to the superior vena cava, and another girl had a coronary artery fistula connecting the left coronary artery with the main pulmonary artery.
A right aortic arch was present in nine of our patients (43%). Although it was more common in children with monosomy 22q11, the difference between the two groups (six of 10 in group I versus three of 11 in group II) did not reach statistical significance (p = 0.14). The presence of an aberrant right subclavian artery (three patients) or left subclavian artery (one patient) was evenly distributed among the patients with monosomy 22q11 and those without (two patients in each group). A high aortic arch was demonstrated in 60% of patients with monosomy 22q11 and in none of those without the deletion (p = 0.004). All patients with a high aortic arch also had a low origin of the innominate artery.
There was no significant difference between the two groups in relation to the presence or absence of confluent central pulmonary arteries (table 2). Confluent central pulmonary arteries were found in eight children in group I and in 10 children in group II. Confluent central pulmonary arteries tended to be smaller in children with monosomy 22q11 than in those without the deletion: the McGoon ratio in group I was 0.8 versus 1.1 in group II. The number of major aortopulmonary collateral arteries did not differ between patients in group I (one to five, mean 3.0) and patients in group II (two to five, mean 3.1).
Pulmonary artery anomalies and outcome
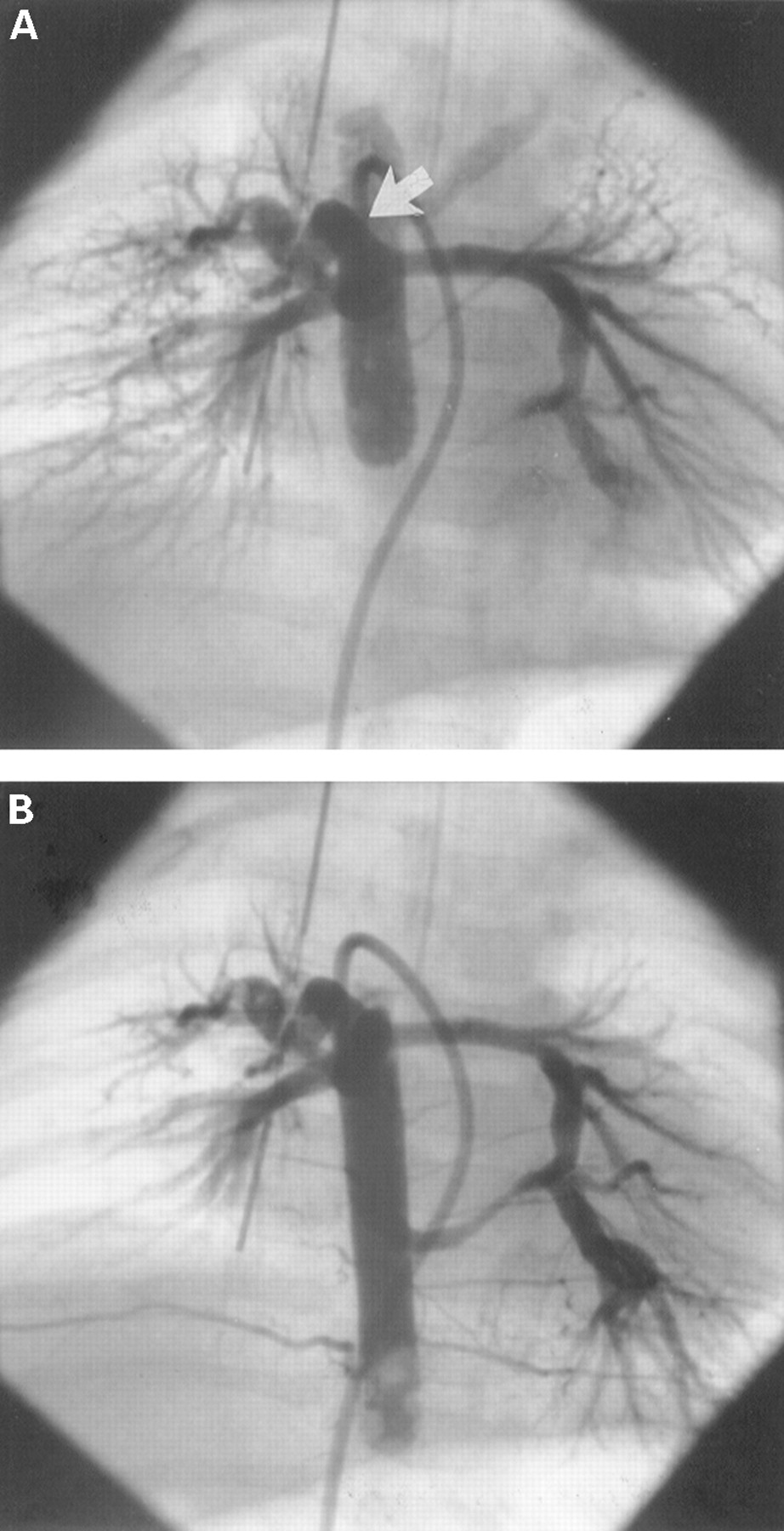
The pulmonary blood supply was unifocal in only one patient in group I, and it was multifocal in the remaining nine children (table2). These latter patients included two children in this group without central pulmonary arteries and seven children who had segments of the pulmonary vascular bed which were perfused by major aortopulmonary collateral arteries without connection to the central pulmonary arteries (fig 3). Complete arborisation of both pulmonary arteries was present in only one patient. Two further patients had complete arborisation of at least one pulmonary artery, while the remaining seven had incomplete arborisation of both pulmonary arteries. Among those patients without monosomy 22q11 (group II), however, the pulmonary blood supply was unifocal in eight children who had complete arborisation of both pulmonary arteries (fig 4). Only three patients in this group had a multifocal pulmonary blood supply with incomplete arborisation of one (two patients) or both pulmonary arteries (one patient). The difference between the two groups with respect to unifocal pulmonary blood supply and the presence of arborisation anomalies was statistically significant (p = 0.006).

Selective injection into a major aortopulmonary collateral artery shows severe intrapulmonary stenoses of left pulmonary artery segments in a patient with monosomy 22q11 (A). Late frames following injection in the descending aorta (B) reveal the presence of severely hypoplastic central pulmonary arteries (arrows).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Unifocal pulmonary blood supply in a patient of group II (A). Balloon blockade angiography in the descending aorta shows filling of the central pulmonary arteries through a large major aortopulmonary collateral artery (arrow). Angiography at a lower level (B) reveals a second major aortopulmonary collateral connecting to the left pulmonary artery, with retrograde filling of the bifurcation (dual blood supply of the central pulmonary arteries).
OUTCOME
At the end of this study successful biventricular repair (including patch closure of the ventricular septal defect and creation of continuity between the right ventricle and the central pulmonary arteries) had not been completed in any of the children in group I (table 2). Palliative procedures have been performed so far in four of these patients. A later biventricular repair seems possible in the patient with a unifocal pulmonary blood supply and in three patients with a multifocal pulmonary blood supply following a successful unifocalisation procedure. The remaining six patients (60%) are not candidates for corrective surgery, because of severe intrapulmonary stenoses (four patients), severe arborisation anomalies (one patient), and associated mitral stenosis (one patient).
Among the patients in group II a successful biventricular repair had been completed at the end of this study in seven children (64%). This included one patient with a multifocal pulmonary blood supply who underwent a successful unifocalisation procedure. Corrective surgery seems likely to be possible in two further patients with unifocal pulmonary blood supply. The remaining two patients with a multifocal pulmonary blood supply are not candidates for a biventricular repair. One of these patients had developed pulmonary hypertension of the left lung due to a large major aortopulmonary collateral artery; the other has severely hypoplastic central pulmonary arteries despite the creation of a large central aortopulmonary shunt.
The difference between the two groups with respect to successful biventricular repair at the end of this study was statistically significant (p = 0.003).
Discussion
The intracardiac anatomy of children with pulmonary atresia and ventricular septal defect is similar to that observed in patients with classical tetralogy of Fallot.14 In contrast to patients with tetralogy of Fallot, there is absence of luminal continuity between the right ventricle and the pulmonary arteries. In the majority of these patients, collateral pulmonary blood flow is provided either by an arterial duct (unilateral or bilateral) or by major aortopulmonary collateral arteries.9 ,10 The possibility of a biventricular repair is determined by the anatomy of the pulmonary arteries. Most patients with collateral lung perfusion provided by an arterial duct have well formed central pulmonary arteries. Hypoplasia or absence of the central pulmonary arteries, arborisation anomalies of the pulmonary vascular bed, or intrapulmonary stenoses are reasons for the reduced feasibility of corrective surgery in patients with collateral lung perfusion provided by major aortopulmonary collateral arteries.9 ,15
Recent studies have shown that monosomy 22q11 is present in a high percentage of patients with pulmonary atresia, ventricular septal defect, and major aortopulmonary collateral arteries.2 The incidence of monosomy 22q11 in this subgroup of patients exceeds that reported among patients with pulmonary atresia, ventricular septal defect, and duct dependent pulmonary circulation, and among patients with classical tetralogy of Fallot without pulmonary atresia.16 ,17 It is noteworthy, however, that all three patients with classical tetralogy of Fallot and major aortopulmonary collateral arteries reported by Momma et al had monosomy 22q11 as well.16 According to these findings the common developmental disturbance in these patients seems not only to produce an abnormality of conotruncal septation but also to have an influence on the development of the derivatives of the sixth aortic arch, which are responsible for the formation of a unilateral or bilateral arterial duct and parts of the central pulmonary arteries. The question arises as to whether this disturbance has an additional influence on the structure of the peripheral pulmonary vascular bed.
Like Digilio et al,1 we found no difference with respect to the presence of confluent central pulmonary arteries between children with monosomy 22q11 and those without, neither was there any difference between the two groups with respect to the number of major aortopulmonary collateral arteries. However, children with monosomy 22q11 had slightly smaller central pulmonary arteries than those without the deletion. A multifocal pulmonary blood supply due to arborisation anomalies of the pulmonary vascular bed was present in a significantly higher proportion of patients with monosomy 22q11 (90% v 27%). Our data suggest that the developmental disturbance caused by monosomy 22q11 impairs the connection of the peripheral pulmonary artery segments to the central pulmonary arteries in patients with pulmonary atresia and major aortopulmonary collateral arteries. Cardiovascular anomalies associated with monosomy 22q11 are attributed to a damage of neural crest cells which play a crucial role in the development of the aortic arches and the conotruncal part of the heart.4 ,18 ,19 Although we are unable to explain the mechanism affecting the disturbed structure of the pulmonary circulation in our patients with pulmonary atresia and ventricular septal defect, these additional pulmonary vascular abnormalities seem to have a major impact on the surgical treatment.
In this retrospective study, the more favourable pulmonary vascular bed in children without the chromosome deletion accounted for the significantly higher proportion of patients undergoing definite biventricular repair (64% in group II versus none in group I). In patients with a unifocal pulmonary blood supply (representing the majority among children without monosomy 22q11), major aortopulmonary collateral arteries can be ligated surgically or occluded at cardiac catheterisation before or at the time of corrective surgery. Since the postoperative right ventricular pressure depends on the number of segments connected to the central pulmonary arteries,20patients with a multifocal pulmonary blood supply (the majority of children with monosomy 22q11) require unifocalisation procedures before corrective surgery. Furthermore peripheral pulmonary artery stenoses, which are often present in these children, may be an additional problem leading to increased right ventricular pressure after a biventricular repair. Although major improvements have been made in the surgical treatment of this extremely complex subgroup of patients,14 ,21 there is no doubt that a many will never be suitable candidates for biventricular repair.22 In well balanced patients with mild symptoms it may even be difficult to decide whether to send them for a unifocalisation procedure or to accept the status quo.15
We are well aware, however, that the retrospective nature of our study imposes limitations. Some patients have died in recent years following surgical procedures or because of the natural course of the disorder, and it is impossible to define retrospectively whether the presence or absence of monosomy 22q11 may have influenced the outcome in these children. The determination of the exact incidence of monosomy 22q11 among patients with pulmonary atresia and ventricular septal defect and its influence on the late outcome will have to be the subject of future prospective studies based on unselected cohorts of patients.