Article Text

Abstract
Objectives: To assess patients with different types of mutations of the β myosin heavy chain (β MHC) gene causing hypertrophic cardiomyopathy (HCM) and to determine the prognosis of patients according to the affected functional domain of β MHC.
Design and setting: Cohort study of subjects referred to an HCM clinic at an academic hospital.
Patients: 70 probands from the HCM clinic were screened for mutations of the β MHC gene and 148 family members of the genotype positive probands were further assessed. The control group for the genetic studies consisted of 106 healthy subjects.
Main outcome measures: Direct DNA sequencing was used to screen 70 probands for mutations of the β MHC gene. Family members underwent genotypic and detailed clinical, ECG, and echocardiographic assessments. The survival of genotype positive subjects was evaluated according to the type of functional domain affected by the missense mutation and according to phenotypic characteristics.
Results: A mutation of the β MHC gene was detected in 15 of 70 probands (21%). Of 148 family members studied in these 15 families, 74 were identified with a β MHC defect. Eleven mutations were detected, including four novel mutations: Ala196Thr, Pro211Leu, Val404Leu, and Arg870Cys. Median survival was 66 years (95% confidence interval (CI) 64 to 77 years) in all affected subjects. There was a significant difference in survival between subjects according to the affected functional domain (p = 0.02). Significant independent predictors of decreased survival were the non-conservative (that is, associated with a change in the amino acid charge) missense mutations that affected the actin binding site (hazard ratio 4.4, 95% CI 1.6 to 11.8; p = 0.003) and those that affected the rod portion of β MHC (hazard ratio 4.8, 95% CI 1.2 to 19.4; p = 0.03). No phenotypic characteristics were associated with decreased survival or cardiovascular morbidity.
Conclusions: The type of β MHC functional domain affected by the missense mutation is predictive of overall prognosis in HCM.
- cardiomyopathy
- genetics
- hypertrophy
- molecular biology
- myosin
- CI, confidence interval
- HCM, hypertrophic cardiomyopathy
- ICD, implantable cardioverter defibrillator
- LV, left ventricular
- MHC, myosin heavy chain
- MLVWT, maximum left ventricular wall thickness
- PCR, polymerase chain reaction
Statistics from Altmetric.com
- CI, confidence interval
- HCM, hypertrophic cardiomyopathy
- ICD, implantable cardioverter defibrillator
- LV, left ventricular
- MHC, myosin heavy chain
- MLVWT, maximum left ventricular wall thickness
- PCR, polymerase chain reaction
Hypertrophic cardiomyopathy (HCM) is a primary and clinically diverse cardiac disorder that is caused by a defect in one of 10 genes encoding proteins of the myofibrillar apparatus: β myosin heavy chain (β MHC), α myosin heavy chain, myosin binding protein C, troponin T, α tropomyosin, troponin I, myosin essential light chain 1, myosin regulatory light chain 2, α cardiac actin, or titin.1–3 There is significant genetic heterogeneity in HCM with more than 150 mutations now implicated in its pathogenesis.1,4,5 The β MHC gene was the first gene identified with this condition.6 Defects of the β MHC gene account for the largest proportion of cases of HCM and many of the initial genotype–phenotype correlative studies of HCM were based on subjects with defects of β MHC.1,4,5,7–9
Certain mutations have been associated with a significantly shorter life expectancy in patients with HCM.4,7–10 However, the associations between specific genotypes and overall prognosis are based on findings from a limited number of families.11 The mutations of β MHC are related to distinct structural and functional domains.12,13 These defects are located in the globular head of myosin (subfragment 1) and cluster at specific locations that are, firstly, associated with actin binding; secondly, close to the nucleotide binding (ATP binding) site; thirdly, adjacent to the region that connects the two reactive cysteine residues; fourthly, at the myosin light chain binding interface; or lastly, at the head–rod junction. We hypothesised that the prognosis of β MHC genotypes is determined by the affected structural–functional domain. We felt that a model based on the various functional domains would be relevant since many loci of β MHC have been identified, the frequency of individual missense mutations is low, and many reported genetic defects are private and novel mutations.14,15 The objectives of this study were to identify mutations of the β MHC gene in patients with HCM and to determine whether the prognosis of these patients may be related to the affected functional domain of β MHC.
METHODS
Study cohort
We identified 70 patients with the diagnosis of HCM who were evaluated in the HCM clinic at the Toronto General Hospital, Toronto, Canada. The initial five subjects were randomly selected from the clinic population and the subsequent 65 probands were consecutive unrelated patients who consented to participate in this study. The relatives of genotype positive probands were contacted and were also studied. The control group for our genetic studies consisted of 106 healthy volunteers. The study protocol was approved by the research ethics board of the Toronto General Hospital.
Identification of positive phenotype
Clinical evaluation consisted of a comprehensive history taking and physical examination, and the review of hospital records, family histories, and necropsy reports. Baseline standard 12 lead ECGs and Doppler echocardiographic studies were performed according to conventional methods.16 Images were obtained from multiple transthoracic windows to assess left ventricular (LV) wall thickness optimally. In addition, we determined the hypertrophy score (range of values 0–10), a semiquantitative echocardiographic method of assessing the extent and severity of LV hypertrophy in patients with HCM.16,17
A positive phenotype in adult subjects was defined as the presence of one of the following diagnostic criteria: firstly, the echocardiographic finding of a maximum LV wall thickness (MLVWT) of ⩾ 13 mm and an increased LV wall to posterior wall ratio of ⩾ 1.316; or secondly, the presence of at least one of the following major ECG abnormalities: voltage evidence of LV hypertrophy (Estes score ⩾ 4)18; abnormal Q waves (defined as ⩾ 0.04 s in duration or > 25% of the amplitude of the ensuing R wave) in at least two contiguous leads; or T wave inversion of ⩾ 3 mm in at least two contiguous leads and in the absence of bundle branch block or hemiblock.19,20 HCM was diagnosed in adolescents (age < 20 years) and children in the presence of a wall thickness of > 2 SD from the mean value (corrected for body surface area).21 ECG and echocardiographic studies were interpreted without knowledge of the genetic status of the study subjects. Deceased relatives whose death preceded the availability of genetic testing at our institution were considered to have been affected with HCM if the condition was identified at necropsy or if death was sudden (defined as non-traumatic death occurring within one hour of onset of symptoms) and premature (age ⩽ 60 years), and there was a definite diagnosis of HCM in a first degree relative, criteria which were similar to those used by other investigators.22
Genetic studies
Genomic DNA was isolated from peripheral blood leucocytes. Exons 3 through 23 of β MHC, encoding the myosin head and head–neck domains, were amplified from genomic DNA with primers designed from intron sequences. Primers flanked the known loci of mutant nucleotide sequences and were based on our previously published sequence of the human cardiac β MHC gene.23,24 Genomic DNA fragments were amplified with the polymerase chain reaction (PCR) using a thermostable TAQ DNA polymerase enzyme and deoxynucleotide triphosphate (Pharmacia, Uppsala, Sweden). PCR products were purified with the QIAquick PCR purification kit (QIAGEN, Santa Clarita, California, USA) to remove residual primers. The amplified PCR products were resolved on a 1% agarose gel (FMC Bioproducts, Rockland, Maine, USA). The Amplicycle (dye terminator) cycle sequencing kit (Perkin-Elmer) was used for sequencing.
β MHC functional sites
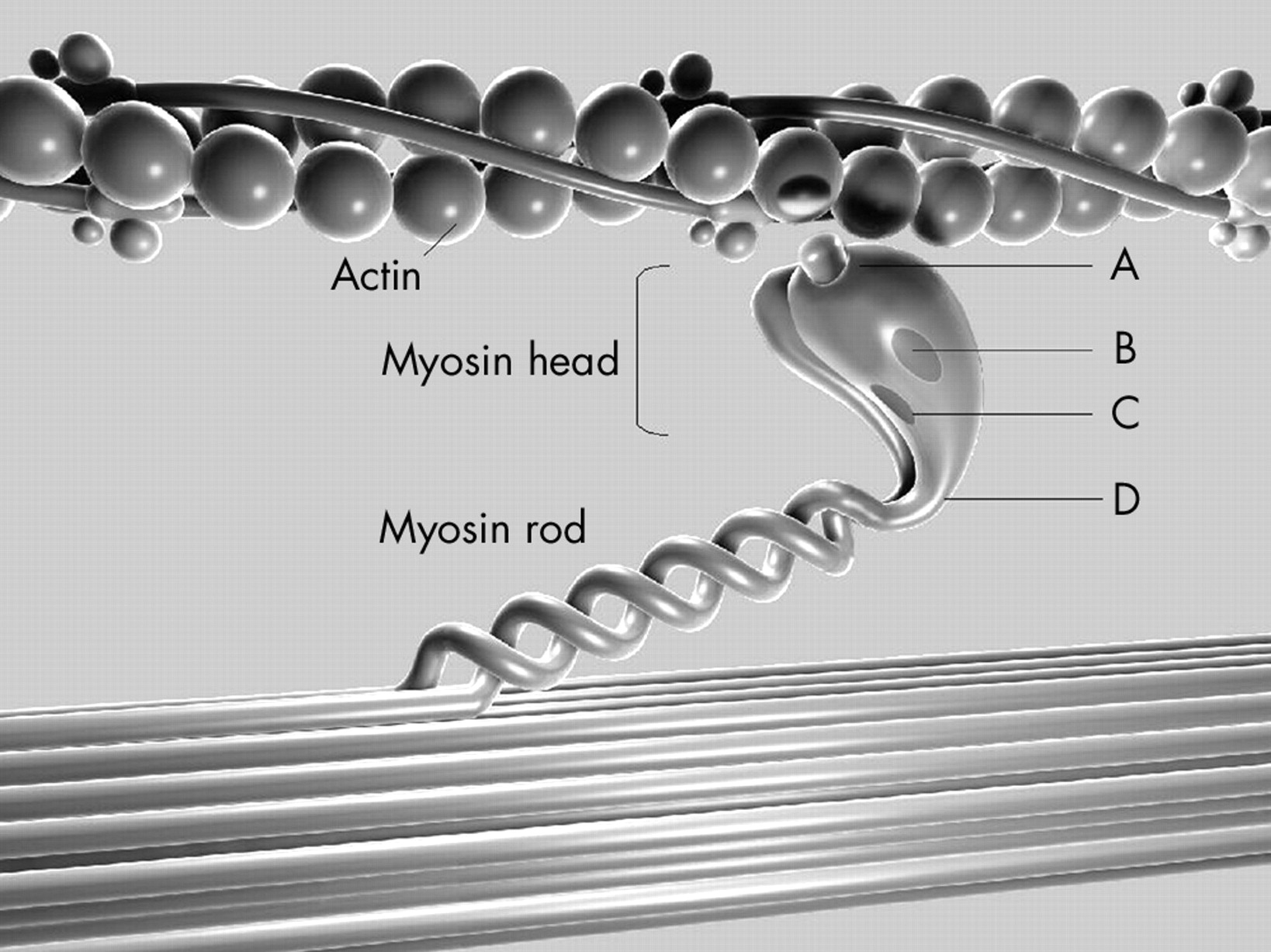
All identified β MHC mutations were categorised based on the location of the mutation in one of the following structural–functional domains: actin binding site, active site (the nucleotide binding pocket and outer end of ATP pocket), essential light chain binding interface, or rod portion (head–rod junction) of β MHC.13 Missense mutations were further subclassified within each functional region based on the presence (non-conserved) or absence (conserved) of a change in the charge of the altered amino acid residue (fig 1).
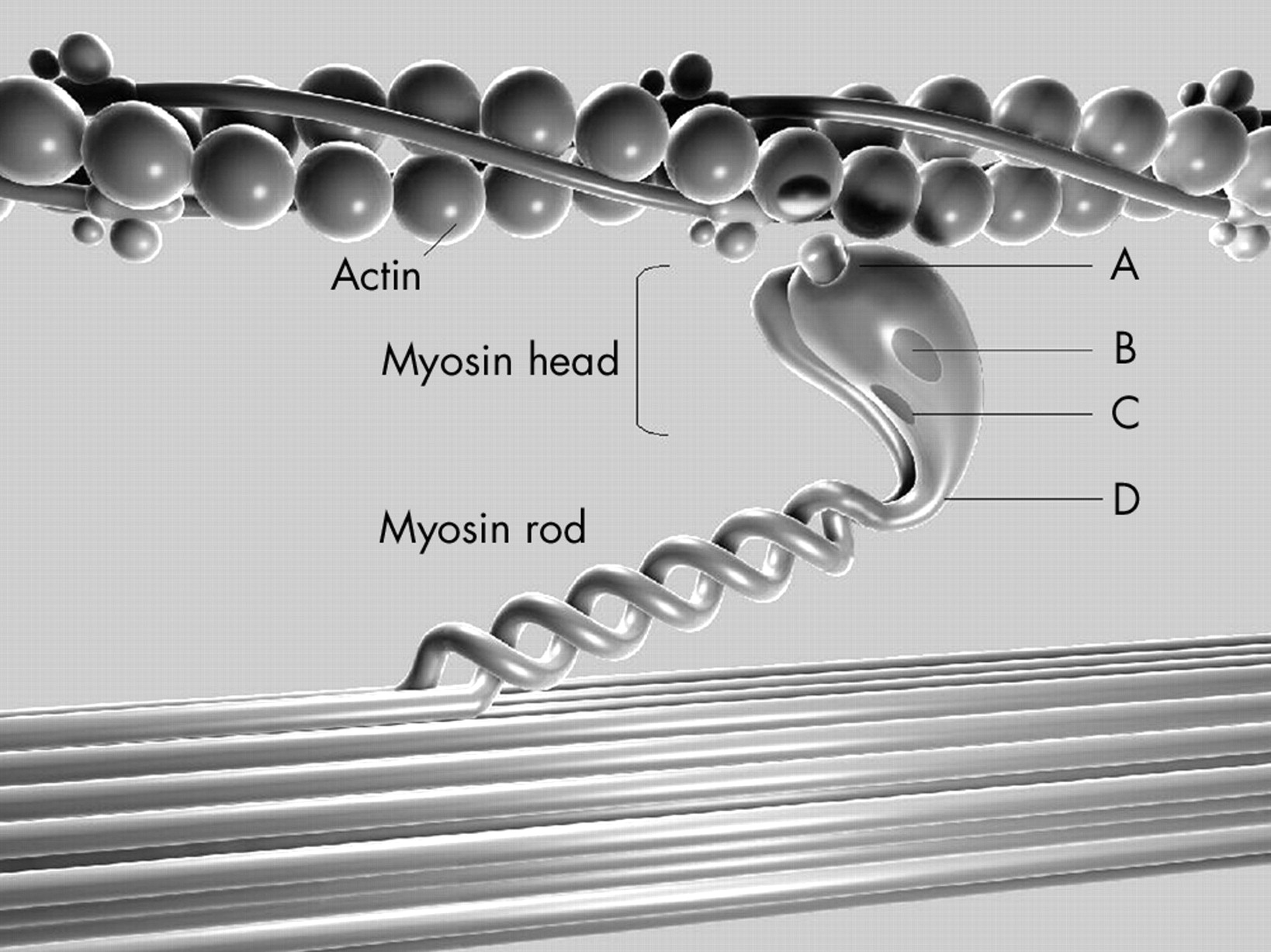
Diagram of the functional domains of the β myosin heavy chain (MHC) and the relation of this sarcomeric protein to actin. The 11 mutations analysed in this study were detected in the following functional domains: (A) actin binding site, (B) active site (ATP binding site), (C) essential light chain binding interface, and (D) head–rod junction. Mutations were further subclassified according to the presence (+) or absence (0) of a change in the charge of the substituted amino acid.
Statistical analysis
Statistical analysis was performed using SAS version 8.0 (SAS Institute Inc, Cary, North Carolina, USA). Continuous data are expressed as mean (SD). Differences between groups were analysed by t tests, analysis of variance, χ2 tests, or Fisher’s exact test, where appropriate. Survival (overall and cardiovascular) was assessed by the Kaplan-Meier method and differences in survival were compared according to the log rank method.25
Multivariate model
Univariate analysis of predictors of death was performed with a Cox logistic regression model. In this study the end point of death was defined as death from any cause, resuscitated cardiac arrest (or appropriate shock from implantable cardioverter defibrillator (ICD)), or cardiac transplantation for end stage HCM. We evaluated the β MHC functional domains and such clinical factors as sex, race, systemic hypertension, and various treatments for HCM as potential predictors. Clinical variables such as history of ventricular tachycardia or a family history of sudden death were not included in the model since we considered these factors to be potential manifestations of the underlying genotype. Univariate predictors with a significance level of < 0.15 were entered into a multivariate Cox proportional hazards model with the use of a backwards elimination algorithm.26 The level of significance for entry into the model was set at 0.05. A secondary analysis was performed in the subgroup of adult subjects for whom baseline echocardiographic data were available. For the latter analysis we evaluated two end points, death or the composite end point of death (as defined above) or serious cardiovascular morbidity (defined as invasive intervention for management of HCM, myocardial infarction or congestive heart failure requiring hospitalisation, stroke, or sustained ventricular tachycardia).
RESULTS
Detection of sequence variants in β MHC gene
Systematic mutation screening of 70 probands identified 11 sequence variants of β MHC in 15 probands (21% of probands studied) (table 1 and fig 1). All 11 mutant β MHC sequences involved a single nucleotide substitution that resulted in an amino acid missense mutation. Additional studies were performed on the relatives of genotype positive probands, with the DNA tested for the specific mutation segregating in their families (fig 2).
β Myosin heavy chain (β MHC) gene mutations identified in study

Pedigrees of four families with hypertrophic cardiomyopathy (HCM) with representative β MHC mutations. Symbols indicate sex and disease status: box, male; circle, female; arrow = proband; darkened = clinical phenotype of HCM; clear = no diagnostic features (clinical, ECG, echocardiography) of HCM; slashed, deceased; HD40, HCM related death at age ⩽ 40 years; HD60, HCM related death at age ⩽ 60 years (deaths include aborted sudden death or cardiac transplantation). Family members were tested for the specific mutation detected in the proband. Plus (+) signs indicate the presence of the genetic defect and minus (−) signs indicate the absence of the mutation. (A) Pedigree of one family with non-conservative actin binding defect, the Arg403Gln mutation. Family is characterised by five premature HCM related deaths (including one early aborted sudden death and one cardiac transplant) and significant cardiovascular morbidity (see text and table 2). (B) Pedigree of family with conservative actin binding defect, the Val404Leu missense mutation. Two premature HCM related deaths have occurred among 12 affected subjects through four generations. (C) Pedigree of family with non-conservative rod defect, the Arg870Cys defect. Three premature deaths (average age at death 52 (7) years) have occurred in this kindred. (D) Pedigree of large kindred with conservative rod defect, the Leu908Val defect. There has been one premature death among 13 affected family members through four generations.
Novel sequence variants
In this group of 15 genotype positive probands we detected four novel abnormal sequence variants that were consistent with disease causing mutations: Ala196Thr (at the active site); Pro211Leu (at the active site); Val404Leu (in actin binding region); and Arg870Cys (in the incipient part of the rod). The evidence that these nucleotide substitutions were responsible for causing HCM was based on five observations. Firstly, each nucleotide alters an encoded amino acid. Secondly, these abnormal sequences occurred in regions that would alter the structure of β MHC. Thirdly, all four sequences encoded amino acids that have been highly conserved in myosin molecules derived from mammalian and rodent species (fig 3), implying functional importance. Fourthly, these mutations have not been found in analyses of 106 normal healthy volunteers (212 chromosomes). Fifthly, the detected sequence variants segregated with all affected patients in the four families studied.

The four identified novel β MHC sequences are highly conserved among other myosin molecules noted in humans and other animals. The identified sequence variants were compared with known sequences of the following myosin molecules: (a) human cardiac β MHC, (b) hamster cardiac β MHC, (c) rat cardiac β MHC, (d) human cardiac α MHC, (e) rat cardiac α MHC, (f) hamster cardiac α MHC, (g) rabbit skeletal MHC, (h) human embryonic skeletal MHC, and (i) rat embryonic skeletal MHC. The numbers above the sequences indicate the amino acid locus of β MHC.
Genetic polymorphisms
In addition, we identified two polymorphic sequence variants in exons 13 and 23 in specimens from three families. One sequence variant (GCC→ACC) resulted in an amino acid substitution of alanine to threonine at locus 393 and the other sequence variant (ATC→GTC) resulted in an amino acid substitution from the isoleucine to the valine residue at locus 909. Both sequences were not detected in any of the samples from control subjects. Further analysis of 35 additional family members determined that these two sequence variants did not cosegregate with subjects who unequivocally had the HCM phenotype, and thus were more consistent with genetic polymorphisms. These three families were excluded from further analysis.
Classification of types of β MHC mutations
We classified β MHC affected subjects into one of six categories (table 2 and fig 1): non-conservative actin binding site mutation (20 subjects); conservative actin binding site mutation (13 subjects); non-conservative active site mutation (5 subjects); conservative active site mutation (6 subjects); non-conservative rod mutation (7 subjects); and conservative rod mutation (21 subjects). We did not analyse the category of defects involving the light chain binding interface since there were only two affected subjects (with the Gly716Arg defect) with a defect involving this functional domain of β MHC in our study group.
Clinical features of subjects with β MHC mutations classified according to genotype and affected domain of β MHC
Summary of clinical findings
By systematic mutation screening of 70 probands we identified 15 kindreds with mutations of the β MHC gene. Evaluation of 148 family members from these 15 kindreds detected 74 subjects affected with these mutations. The mean age at diagnosis of affected subjects was 41 (21) years and 51% were men. Affected adult subjects had an ECG derived Estes score of 3 (2) (range 0–8), and echocardiographic studies showed a MLVWT of 18 (7) mm (range 7–33 mm) and an echocardiographic hypertrophy score of 4 (3) (range 0–10). Ten genotype positive adults had normal LV wall thickness. In these 10 subjects, the detection of an abnormal β MHC genotype clarified the diagnosis in four genotype positive subjects who had positive ECG but negative echocardiographic criteria for the diagnosis of HCM, and established the diagnosis in six genotype positive young adults (all 35 years old or less at the time of evaluation) who fulfilled neither ECG nor echocardiographic criteria for HCM. There were no significant differences in the presence of LV hypertrophy, the hypertrophy score, the MLVWT, or the left atrial size among subjects categorised according to the β MHC functional domains. Table 2 summarises additional phenotypic characteristics of the identified mutations.
Predictors of survival
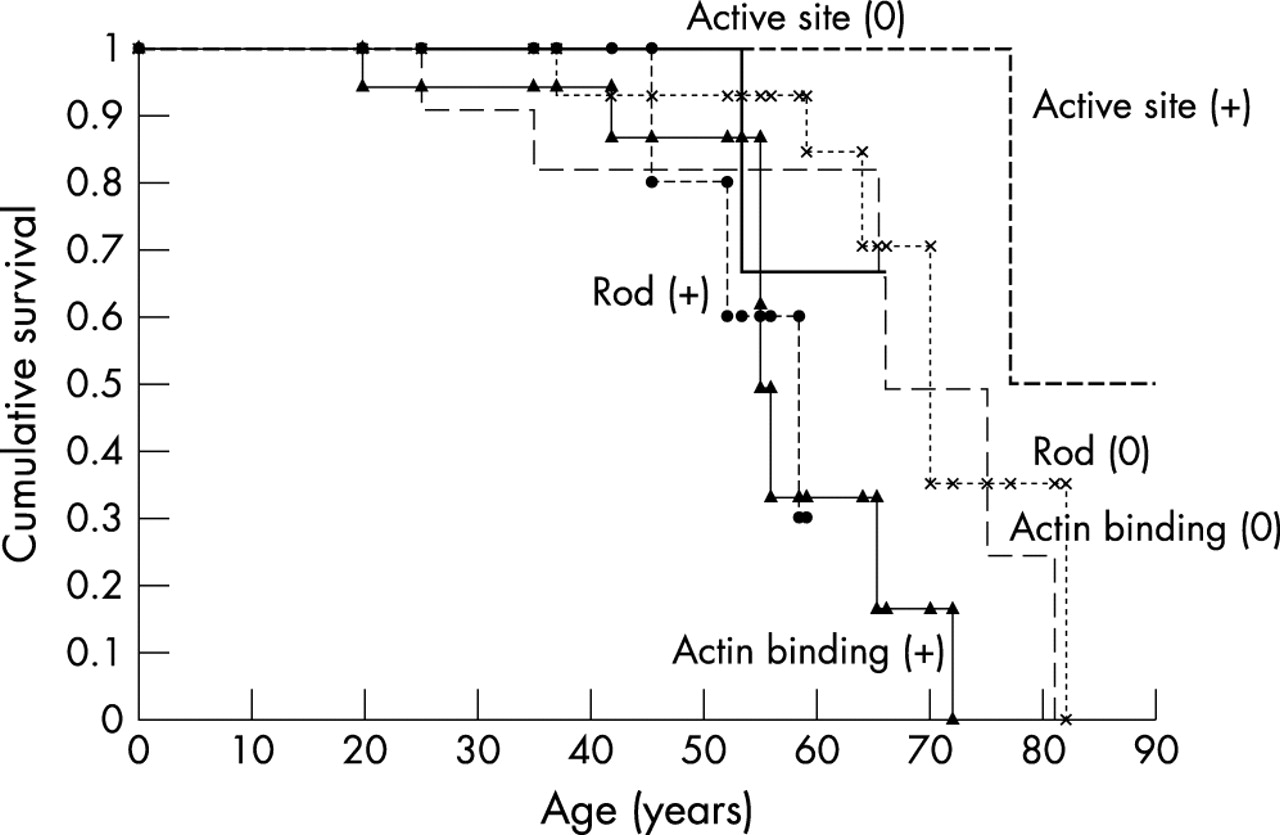
Overall median survival of all affected subjects was 66 years (95% confidence interval (CI) 64 to 77 years). There was a significant difference in overall survival (p = 0.02) (fig 4) and in cardiovascular survival (p = 0.02) between the subjects categorised according to the six functional domains. There were two non-cardiovascular deaths, secondary to leukemia in one patient with the Arg249Gln defect and hepatic malignancy in one patient with the Val404Leu defect. On the basis of the multivariate Cox logistic regression model the only independent predictors of mortality in this study were the β MHC functional domains of a non-conservative actin binding defect (hazard ratio 4.4, 95% CI 1.6 to 11.8; p = 0.003) and a non-conservative head–rod junction defect (hazard ratio 4.8, 95% CI 1.2 to 19.4; p = 0.03). The specific missense mutations affecting these functional domains were the Arg403Gln, Arg870Cys, and Glu930Lys defects. The survival of subjects affected by any of these three missense mutations was significantly reduced compared with that of the other subjects in the study cohort (p = 0.003).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier (product limit) curves for the survival of patients with β MHC mutations in the study population. There was a significant difference in survival between subjects classified according to the affected functional domain and the charge change of the encoded amino acid (p=0.02, log rank test). Survival was significantly shortened in subjects with actin binding (+) or rod (+) defects. (0) indicates conservative amino acid substitution, (+) indicates non-conservative amino acid substitution. (1) Actin binding (0) defects: (— — —); (2) actin binding (+) defects: (▴—▴); (3) active site (0) defects: (); (4) active site (+) defects: (- - - - -); (5) rod (0) defects: (x..........x); and (6) rod (+) defects (• - - - - - •).
In one family of 14 affected members with the Arg403Gln defect involving the actin binding site, there was a history of aborted sudden death in a 19 year old man, a cardiac transplant because of end stage HCM at age 30 in his father, surgical myectomy required in three family members, and sudden death before the age of 60 in three additional relatives (fig 2A). The second family with the Arg403Gln defect had six affected members, including one family member who underwent myectomy at age 15, one who developed recurrent ventricular tachycardia and required an ICD at age 42, and two who died before age 60 from their underlying HCM. In the other three families affected by the Arg870Cys or Glu930Lys mutations, there were three premature cardiac deaths and two patients requiring myectomy. In contrast, there were two premature deaths among 21 affected subjects (in three families) with the Leu908Val non-conservative head–rod junction defect. There were no significant differences in the cardiovascular event rates when we stratified patients according to phenotypic characteristics: (1) no echocardiographic evidence of disease penetrance (MLVWT < 12 mm) versus MLVWT ⩾ 13 mm; (2) MLVWT < 20 mm versus MLVWT ⩾ 20>mm; or (3) a low (score of < 5) or high (score of ⩾ 5) echocardiographic hypertrophy score.
DISCUSSION
Summary of study findings
By systematic mutation screening of 70 subjects, the majority of whom were part of a consecutive series of patients undergoing genotyping, 11 disease causing sequence variants of the β MHC gene were identified. There was striking genotypic heterogeneity in our cohort, with 11 different missense mutations detected in 15 unrelated kindreds. This group included four newly identified missense mutations. Further testing of 148 family members determined that prognosis in affected family members could be determined by the underlying affected β MHC functional domain. Survival was significantly shortened in subjects with non-conservative missense mutations affecting the actin binding site or the head–rod portion of β MHC.
Predictors of risk with β MHC mutations
Our model of assessing risk in subjects with β MHC defects based on the affected functional domain had a number of advantages. Our study incorporated the findings of a wide range of pedigrees, ranging from small families to larger kindreds, and the findings from 11 genotypes. The prognosis of individual genotypes has previously largely been derived from studies of highly selected large pedigrees. The classification of individual missense mutations into different functional domains permitted the analysis of multiple genotypes and provided a framework for the assessment of novel mutations. We also assessed non-genetic but potentially important modifying or environmental variables such as sex, race, history of systemic hypertension, and use of medications. Our study suggests that the underlying genotype is more predictive of outcome than these non-genetic factors.
Furthermore, this study cohort differed from cohorts in two recent studies that examined the prevalence of 13 mutations from the genes of three sarcomeric proteins (β MHC, troponin T, and α tropomyosin) in 293 unrelated subjects referred to a tertiary referral centre.10,27 These mutations were classified as “malignant” or “benign” based on prior published studies.4,5,7,8,11,22 Ten missense mutations of β MHC were evaluated in the two studies. The malignant β MHC mutations were four non-conservative missense mutations involving the actin binding site, the active site, and the light chain binding interface. The benign β MHC mutations were six defects that did not result in non-conservative mutations of the two critical functional domains identified in our study, the actin binding site or the head–rod junction. Both studies found a low prevalence of these 10 β MHC mutations (two patients (0.7%) with a malignant β MHC defect and five patients (1.7%) with a benign β MHC defect). The seven subjects identified with these β MHC mutations had different clinical manifestations of HCM and variable family histories. Although the five subjects with the benign β MHC mutations had all experienced major cardiovascular events, the entire study cohort of 293 subjects appeared to have a significant burden of disease with 32% reporting a family history of HCM and 28% undergoing myectomy.10,27 The studies were cross sectional in design, did not systematically examine pedigree data, and identified only a small number of genotype positive subjects, which precluded an analysis of the differences in survival and longitudinal outcomes between the types of mutations. The frequency of these 10 particular β MHC mutations in our own study cohort would be 5.7% (4 of 70 probands) of the malignant β MHC defects and 4.3% (3 of 70) of the benign β MHC defects.
Critical functional sites of β MHC
This analysis based on the affected β MHC functional domain complements previous genotype–phenotype correlation studies. Missense mutations of β MHC that result in an amino acid charge change, presumably leading to conformational structural changes and functional abnormalities of the sarcomeric protein, have been associated with a worsened prognosis.7,9,11 The non-conservative missense mutations, Arg403Gln (in actin binding region) and Arg719Cys (light chain binding interface), have been characterised by high disease penetrance, a high incidence of sudden cardiac death, and a decreased life expectancy. In contrast, patients with the Leu908Val defect, a conservative mutation occurring in the head–rod junction, are characterised by a low incidence of sudden death and a more benign course.7,9,28 The presence of a non-conserved mutation in critical regions probably further disrupts the ambient environment and the function of β MHC.
Structural and functional alterations of myosin
Myosin hydrolyses ATP and undergoes conformational changes initiated in the globular head and transmitted to the rod domain. These actions lead to sliding of the myofilaments of the sarcomeric apparatus. Missense mutations of the β MHC gene affect different structural and functional domains of the head or the head–rod junction of the β MHC protein. These mutations appear to cause the incorporation of abnormal polypeptides, resulting in contractile dysfunction and an alteration in sarcomeric and myocyte structure and organisation.29 The non-conservative missense mutations affecting the actin binding and rod regions imparted the worse prognosis, suggesting that derangements to the actin–myosin interaction and the transmission of force to the thick filament, respectively, would lead to a particularly malignant HCM phenotype.13
Other studies have provided additional insight into the pathogenesis of HCM caused by the disruption of the actin binding site by the Arg403Gln missense mutation. In a transgenic mouse model of the Arg403Gln defect, the mechanical coordination between the two heads of cardiac myosin was perturbed, which may result in abnormal power output and which is a potential stimulus for the hypertrophic response.30 Another study has suggested that this defect results in abnormal kinetics and is not caused by a change in the mechanical properties of the myosin motor.31 Regardless of the primary mechanism, a mutation in this functionally critical region disrupts the actin–myosin interface and causes overt phenotypic manifestations. One study detected labile temporal repolarisation, as quantified by QT variability analysis, in patients with the Arg403Gln mutation.32 In addition, reduced myocardial tissue Doppler velocities, reflective of myocardial dysfunction, has been found in a transgenic rabbit model of HCM.33
Genetic heterogeneity of HCM
The prevalence of mutations of the β MHC gene was 21% in the probands in our study. Various studies have detected mutations of the β MHC gene in 6–48% of probands evaluated.5,7,34,35 This wide range in the reported prevalence is probably caused by differences in the study populations, differences in the study protocols, which may have included the evaluation of other sarcomeric proteins,34 and differences in the methods used to identify mutations of the β MHC gene.1,7,34,35
There may be gene specific variations in the clinical and morphologic expression of phenotype positive patients. Patients with troponin T mutations may have decreased disease penetrance and a milder degree of hypertrophy, but a significantly higher risk of sudden death than patients with certain β MHC missense mutations.22 Mutations of the cardiac myosin binding protein C gene have been associated with a milder form of hypertrophy and delayed phenotypic expression until middle or older age.36,37 The distribution of mutations responsible for HCM of the elderly is distinctly different from that of mutations associated with familial early onset HCM.15
The era of molecular medicine has generated new dilemmas in the screening, diagnosis, and treatment of HCM.9 Risk stratification for the development of sudden death in HCM is challenging and controversial, and has relied predominantly on the assessment of phenotypic findings.38 Recent studies of screening for recognised mutations in unrelated subjects underscore the low yield and difficulties in widespread genetic testing.10,27 Additional concerns that need to be considered in the use of molecular genetic testing in the management of patients with HCM are the more widespread use of ICDs in this condition39 and the development of multiple treatment strategies aimed at relieving disease associated symptoms.3
Limitations of the study
Baseline clinical characteristics were collected by retrospective review of medical records. There may have been recall bias in those families in which there had been premature deaths. The occurrence of malignant events in family members may influence other family members’ decisions to seek medical attention, serial screening, and follow up. Furthermore, a family history of sudden death may have had a significant impact on physicians’ advice, treatment decisions, and vigilance in the ongoing monitoring and management of patients and their family members. Our multivariate model did not consider all structural–functional domains of β MHC since an insufficient number of patients with mutations affecting certain β MHC regions was identified in our study group. Finally, we did not assess other factors in our study, such as modifier genes or trophic factors, which have been shown to modify the phenotypic expression of HCM.40,41
Conclusions
DNA sequencing of the β MHC gene permitted the detection of mutant β MHC genotypes in 21% of unrelated probands. The results of our study suggest that there are functionally critical regions of β MHC and that mutations involving these sites portend a significantly worsened prognosis. Non-conservative missense mutations involving the actin binding site and the rod portion of β MHC are independent predictors of an adverse outcome. The more widespread application of molecular genetic analyses will greatly enhance the ability of investigators to optimise the risk stratification and management of patients with HCM.
Acknowledgments
This work was supported by the Hypertrophic Cardiomyopathy Research Fund at the University of Toronto and by a grant from the Heart and Stroke Foundation of Canada.