

Abstract
Sodium currents were recorded from motoneurons that were isolated from mice at postnatal days 0–8 (P0–P8) and maintained in culture for 12–24 hr. Motoneurons from normal mice exhibited a more than threefold increase in peak sodium current density from P0 to P8. For mice lacking a functional Scn8a sodium channel gene, motoneuronal sodium current density was comparable at P0 to that of normal mice but failed to increase from P0 to P8. The absence of Scn8asodium channels is associated with the phenotype “motor end plate disease,” which is characterized by a progressive neuromuscular failure and is fatal by 3–4 postnatal weeks. Thus, it appears that the development and function of mature motoneurons depends on the postnatal induction of Scn8a expression.
- motoneurons
- sodium channels
- Scn8a
- postnatal development
- motor end plate disease
- neuromuscular system
- mouse
Motoneuronal electrical activity plays a critical role in the prenatal and postnatal development of the neuromuscular system (for review, see Grinnell, 1995). For example, individual muscle fibers are multiply innervated at birth but are singly innervated in the adult. Elimination of the supernumerary synapses occurs during the first 2 postnatal weeks and is governed by neuromuscular activity, which is obviously influenced by the ion channels expressed in the motoneurons. Previous work has shown that the expression of calcium currents in motoneurons changes during the period of initial synapse elimination (Mynlieff and Beam, 1992b). Sodium current density has been shown to increase as a function of the length of time that embryonic mouse motoneurons are maintained in vitro (MacDermott and Westbrook, 1986). However, the in vivo postnatal development of motoneuronal sodium currents has not been characterized. Here, we have measured sodium currents in identified motoneurons that were obtained from mice of varying postnatal ages and maintained overnight in primary culture.
In addition to cells from normal mice, we examined motoneurons from mice with motor end plate disease (med), a genetic defect that causes progressive neuromuscular failure. The gene altered by themed mutation was recently identified (Burgess et al., 1995) as the voltage-gated sodium channel Scn8a that is expressed in brain and spinal cord (Schaller et al., 1995). Four independent mutations of Scn8a have been characterized at the molecular level: a transgene-induced intragenic deletion (Burgess et al., 1995), two mutations that affect splicing (Kohrman et al., 1996a), and one missense mutation (Kohrman et al., 1996b). Mice homozygous for the transgene-induced mutation lack functional Scn8a channels and display altered cerebellar function and progressive neuromuscular weakness, which begins ∼10 d after birth and results in death within 3–4 weeks (Burgess et al., 1995). Analysis of homozygous transgenic mice demonstrated that Scn8a channels play an important role in spontaneous and repetitive firing of cerebellar Purkinje cells (Raman et al., 1997). Mice homozygous for the missense mutation display profound cerebellar dysfunction without neuromuscular weakness and survive to adulthood (Dick et al., 1986; Harris and Pollard, 1986). Early studies of mice homozygous for the splicing mutations demonstrated progressive loss of evoked neurotransmitter release at the neuromuscular junction (Duchen and Stefani, 1971; Weinstein, 1980;Harris and Pollard, 1986), indicating that the Scn8a channel is essential for motoneuron function. Scn8a transcripts have been detected in motoneurons of the rat by in situhybridization (Schaller et al., 1995; Felts et al., 1997). However, no previous studies have evaluated the quantitative contribution ofScn8a to motoneuronal sodium current.
We found that peak sodium current density in motoneurons from normal mice increased more than threefold between postnatal day 0 (P0) and P8. At P0, motoneurons from mice homozygous for a disruptedScn8a gene had a sodium current density that differed little from that of normal P0 motoneurons. However, sodium current density failed to increase between P0 and P8 in motoneurons lacking an operational Scn8a gene. Thus, the postnatal increase in sodium current expression that occurs in normal motoneurons depends on an intact Scn8a gene and appears to be essential for the development and function of mature motoneurons.
MATERIALS AND METHODS
Animals. The medtg mutation arose by nontargeted insertion of a transgene into a chromosome derived from strain C3 Hr/HeJ mice and has been maintained by crossing with strain C57BL/6J since 1992 (Kohrman et al., 1995). C57BL/6J mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Heterozygous transgenic mice (genotype tg/+) were crossed to produce litters containing +/+, tg/+, and tg/tg mice. See Figure1 for control studies using strain 129/ReJ mice (Jackson).
Motoneuron cultures. The procedures used for culturing motoneurons were similar to those described previously (Mynlieff and Beam 1992a,b; Garcı́a and Beam, 1996). Neonatal mouse pups were anesthetized and injected in all four limbs with a suspension of DiI (Molecular Probes, Eugene, OR) (2.5 mg/ml, 20% ethanol, 80% rodent Ringer’s solution with 0.1% bovine serum albumin). The pups were returned to their mothers for 9–11 hr to allow the dye to label motoneuronal cell bodies (Honig and Hume, 1986). The pups were then reanesthetized and decapitated, and the spinal cords were removed in oxygenated rodent Ringer’s solution (in mm: 146 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 11 glucose, and 10 HEPES, pH 7.4). The spinal cord was cleaned of meninges and any adhering dorsal root ganglia, cut into small pieces (<1 mm3), and placed in 0.5 ml of a 0.1% type XI trypsin and 0.01% DNase I (both from Sigma, St. Louis, MO) solution in PIPES-buffered saline (in mm: 120 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 25 glucose, and 20 piperazine-N,N′-bis[2-ethanesulfonic acid], pH 7.0). After 15–20 min of incubation at 35°C, the tissue was rinsed with neural basal medium containing B27 supplement (Life Technologies, Grand Island, NY), 100 μg/ml streptomycin, and 60 μg/ml penicillin, and triturated with a fire-polished pipette. The cells were plated onto 35 mm poly-l-lysine-coated (4–15 kDa; 1 mg/ml in 0.15m boric acid, pH 8.4) dishes, maintained overnight in a humidified atmosphere of 95% air and 5% C02 at 37°C, and recorded from on the following morning.
Ionic currents. The majority of recordings (>75%) were obtained from motoneurons identified on the basis of DiI staining (Honig and Hume, 1986), with the remainder from nonlabeled cells identified as motoneurons on the basis of morphological criteria (Smith et al., 1986; Milligan et al., 1994; Mynlieff and Beam, 1994). Current densities in nonlabeled cells were not significantly different from those in labeled cells (see Results). Membrane currents were recorded at room temperature (20°C) with whole-cell patch-clamp configuration (Hamill et al., 1981) using a DAGAN 3900 patch-clamp amplifier (Dagan, Minneapolis, MN) equipped with a 3911 whole-cell expander (Dagan). The currents were electronically filtered at 1 kHz (8-pole Bessel filter) and then sampled and stored with a digital computer (Indec Systems, Capitola, CA). Linear components of leak and capacitive currents were removed from test currents by digital subtraction of scaled control currents elicited by 20 mV hyperpolarizations from the holding potential (−80 mV). To normalize for differences in membrane area, current densities were calculated as the ratio of measured current to linear cell capacitance. Data are given as mean ± SEM. Data were tested for significant differences (p < 0.05) by an unpaired Student’s t test.
Patch electrodes were made from soda lime glass, had resistances of 4–5 MΩ, and were coated with wax to reduce capacitance. The pipette filling solution contained (in mm): 140 Cs-aspartate, 5 MgCl2, 10 Cs2EGTA, and 10 HEPES, pH 7.4. For measurement of sodium currents, the bath contained (in mm): 35 NaCl, 115 TEA-Cl, 2 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.4. For measurement of calcium currents, the external solution contained (in mm): 10 CaCl2, 145 TEA-Cl, 0.0005 TTX, and 10 HEPES, pH 7.4.
Ribonuclease protection assay. Spinal cords from wild-type (+/+) mice at P0 (n = 6), P7 (n = 5), and adult (n = 3) were pooled, and total RNA was prepared using the Trizol reagent (Life Technologies). Ribonuclease protection assays were performed as described previously, using a 511 bp [α-32P]UTP-radiolabeled riboprobe that contained a 381 bp coding fragment (Burgess et al., 1995).
Determination of genotype. Genotype was determined either from muscle samples obtained at the time of spinal cord dissection or from toe clips obtained at P3 or P4. The samples were frozen on dry ice and stored at −80°C until further processing. Genomic DNA was prepared from the muscle samples by digestion with proteinase K, phenol–chloroform extraction and ethanol precipitation, and prepared from the toe clips by a modification of the method of Pomp and Murray (1991). Specifically, these samples were digested for 2 hr at 55°C in 250 μl of PCR buffer (10 mm Tris-HCl, pH 8.3, 50 mm KCl, 0.01% gelatin, and 1.5 mmMgCl2) containing 0.45% Tween 20, 0.45% NP-40, and 0.2 μg/μl proteinase K. Five-microliter aliquots were incubated under mineral oil for 10 min at 95°C to inactivate proteinase K and amplified in 25 μl PCR using the multiplexed PCR protocol described below.
PCR reactions contained 50–100 ng of genomic DNA. Two genotype assays were used. One was a previously described assay involving separate amplification of the transgene and the wild-type Scn8aallele (Raman et al., 1997). In the new multiplex assay, a single aliquot of DNA from each mouse was amplified with two compatible primer pairs: primer pair 1 (forward, GAGGG AGGGC TGAGG GTTTG AAGTC; reverse, CCATG GTGTC TGTTT GAGGT TGCTAG) amplifies a 244 bp fragment from the transgene (Kohrman et al., 1995); and primer pair 2 (forward, GAAGT GAAAC CTTTA GAGGA GCTGT ATGAG; reverse, GGAAT TCCTT CTGGA AGTCG CCGTT CCTGT GAATG TCC) amplifies a 103 bp fragment of exon 14 from the wild-type Scn8a that is missing in the transgenic allele (Raman et al., 1997). After denaturation for 3 min at 94°C, PCRs were performed for 35 cycles with annealing for 45 sec at 65°C, followed by 45 sec at 72°C and 45 sec at 94°C.
RESULTS
Postnatal increase in sodium current density in normal motoneurons
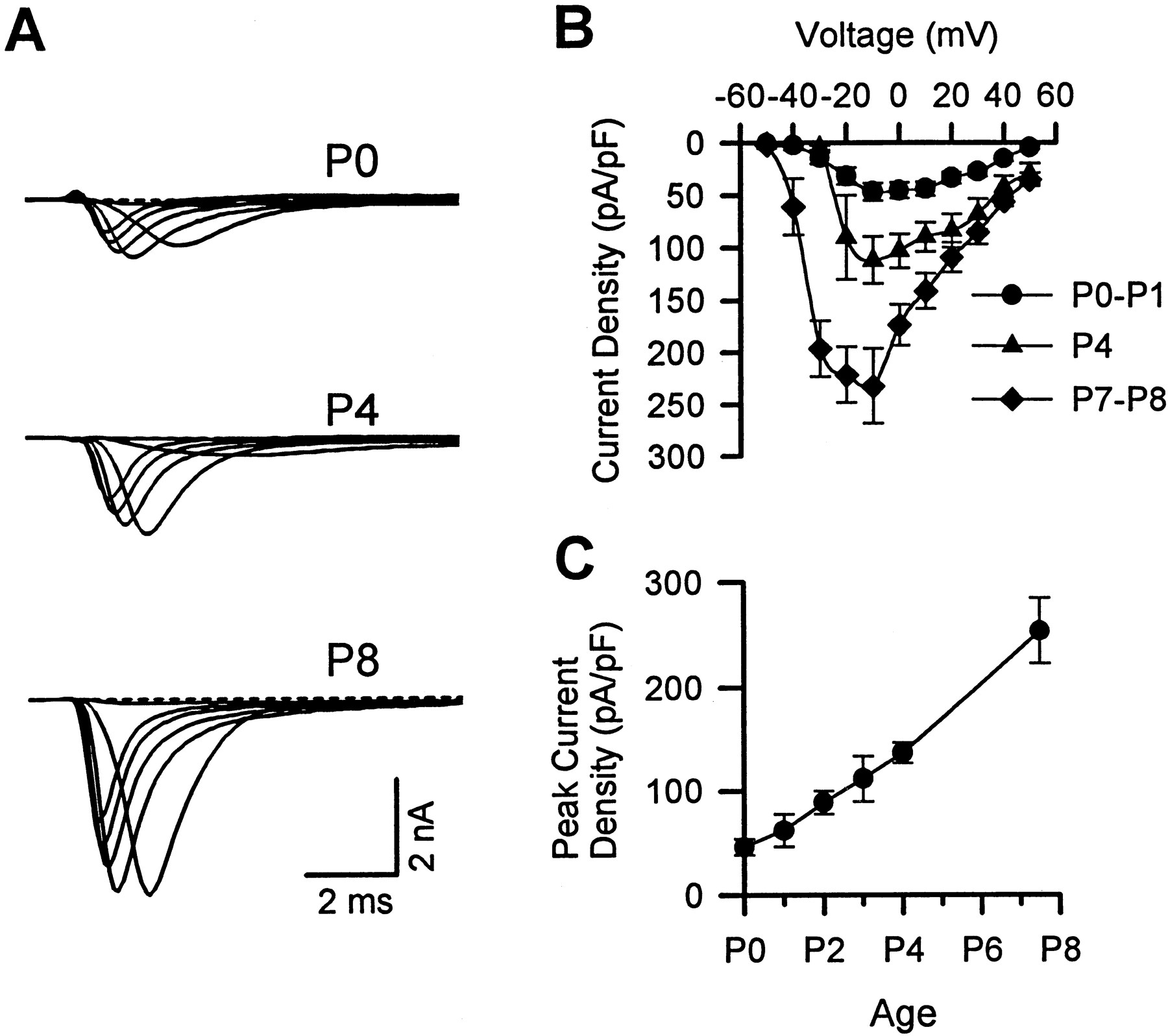
Figure 1Aillustrates representative sodium currents in motoneurons isolated from P0, P4, and P8 mice and cultured overnight. It is evident that the P4 currents are larger than the P0 currents, and that the P8 currents are substantially larger still. This postnatal increase in sodium current is also apparent in the I–V relationships obtained by averaging data obtained from many cells (Fig. 1B). Measured at a test potential of −10 mV, peak current density increased almost fivefold between P0 and P7–P8 (Fig. 1C). As can be seen in the raw currents, the high-resistance pipettes necessary to record from motoneurons often caused a loss of voltage control for potentials in the negative slope region of the I–V curve. This was true, although the currents were measured with a reduced extracellular sodium concentration (35 mm). Thus, it is difficult to determine whether there were developmental changes in either voltage dependence or kinetics.

Postnatal increase of sodium current density in normal mouse motoneurons. A, Representative sodium currents in motoneurons isolated from normal mice at P0, P4, and P8 and cultured overnight. Here (and in Fig. 4), the illustrated currents were elicited by step depolarizations ranging from −40 to +10 mV at 10 mV intervals, extracellular sodium concentration was 35 mm, and the holding potential was −80 mV. B, Average current–voltage relationships for motoneurons obtained from normal mice at P0–P1 (n = 45), P4 (n= 7), and P7–P8 (n = 12). Error bars indicate ± SEM. C, Peak sodium current density as a function of postnatal age. The data point plotted midway between P7 and P8 was obtained from P7 and P8 motoneurons (n = 28, 17, 16, 9, 8, and 12 at P0, P1, P2, P3, P4, and P7–P8, respectively).
Scn8a transcripts in neonatal spinal cord
Previous analysis with a specific ribonuclease protection assay demonstrated that the Scn8a transcript is present in the spinal cord of normal adult mice but absent from the spinal cord of homozygous transgenic mice lacking a functional Scn8a gene (Burgess et al., 1995). To determine whether the Scn8atranscript is present during early postnatal development of the normal spinal cord, RNA was prepared from spinal cords of mice at P0 and P7. The Scn8a transcript was readily detected at both P0 and P7 (Fig. 2), consistent with the hypothesis that Scn8a channels contribute to the postnatal increase in motoneuronal sodium current illustrated in Figure 1.

Expression of Scn8a in mouse spinal cord. Twenty-microgram aliquots of total RNA were assayed by ribonuclease protection using a 511 bp Scn8a specific riboprobe. The predicted size of the protected fragment is 381 bp.Left panel, All samples were run on the same gel, which was dried and exposed to film for 1.5 hr (probe) or 5 hr (samples plus probe). Right panel, The concentration and integrity of the RNA samples were compared by staining with ethidium bromide after electrophoresis of 1 μg aliquots on a 1.0% agarose gel.
Identification of mice with a disrupted Scn8a gene
To evaluate the contribution of Scn8a to the postnatal increase in motoneuronal sodium current, we examined cells from homozygous medtg mice in which both copies of theScn8a gene are inactivated by transgene (tg) insertion. Homozygous mice were obtained from crosses betweentg/+ heterozygotes. To determine genotypes of the offspring, genomic DNA from each animal was analyzed by PCR with two primer pairs: one for amplification of the transgene and one for amplification of the wild-type Scn8a gene. Genotypes are inferred from the presence or absence of transgenic and wild-type allele products of 244 and 103 bp, respectively (Fig. 3).

Determination of Scn8a genotypes by PCR. Genomic DNA was prepared from offspring of the cross (tg/+ × tg/+) and amplified using two pairs of primers, as described in Materials and Methods. Amplification of the wild-type Scn8a gene generated a 103 bp product; amplification of the transgene generated a 244 bp product. The inferred genotypes, based on presence or absence of each product, are shown above each lane. A minor 280 bp product was occasionally amplified from wild-type DNA by the transgene primers (arrowhead). The positions of molecular weight markers (1 kb ladder; BRL, Bethesda, MD) are shown at the leftin base pairs.
Sodium currents in motoneurons frommedtg mice
To determine whether the absence of a functional Scn8agene affected motoneuronal sodium currents, cells were isolated from P0–P8 mouse pups obtained from tg/+ crosses. For each pup, a sample of tissue was frozen at the time of spinal cord dissection. Mouse genotype was determined by PCR analysis only after currents had been recorded and analyzed. This, together with the fact that P0–P8medtg mice do not exhibit any phenotypic signs of altered neuromuscular function, ensured that the currents were recorded under blind conditions.
As shown by both raw currents (Fig.4A) and averageI–V relationships (Fig.4B–D), sodium currents were nearly normal in motoneurons from P0–P3 medtg mice but very much reduced in cells from P7–P8 medtg mice. Of 11 P7 and P8 medtg motoneurons analyzed, 5 had little or no sodium current (peak density of ≤15 pA/pF) like the one illustrated in Figure 4A, 4 had modest sodium currents (peak density of ∼45 pA/pF), and 2 had currents (peak densities of 91 and 142 pA/pF) almost as large as the mean for normal cells. This variability may reflect the biological heterogeneity of the motoneuron pool, because the neuromuscular weakness characteristic of motor end plate disease is not uniform from muscle to muscle (Duchen, 1970;Duchen and Stefani, 1971).

Lack of postnatal increase in sodium current density in medtg motoneurons. A, Representative sodium currents in motoneurons isolated frommedtg mice at P0, P3, and P7. Note that the currents from the P7 motoneuron are displayed at a 10-fold higher gain.B, C, D, Average current–voltage relationships for wild-type andmedtg motoneurons at P0–P1, P2–P3, and P7–P8. In B, C, and D,n = 4, 7, and 11 for the wild-type motoneurons, andn = 6, 9, and 11 for themedtg motoneurons, respectively. Each of the data points is based on currents measured in at least two separate motoneuron cultures.
For the P7 medtg motoneuron illustrated in Figure4A, the sodium current was so small that it was partially obscured by another current, with an amplitude and voltage dependence suggestive of calcium current (the bath contained 2 mm calcium). Similar presumptive calcium currents were seen in other P7–P8 medtg cells with very small sodium currents, indicating that the small sodium currents were not attributable to a generalized membrane pathology. We also recorded currents from a few medtg cells under conditions that isolate calcium currents (sodium-free external solutions with 10 mm calcium and 0.5 μm tetrodotoxin). Currents recorded from medtg cells under these conditions (Fig. 5) were qualitatively similar to previously described calcium currents in normal motoneurons (Mynlieff and Beam, 1992a,b; Garcı́a and Beam, 1996).

Calcium currents in motoneurons from a P0 (top traces) or a P6 (bottom traces)medtg mouse. Test pulses were to potentials of −20 to +20 mV, in 10 mV increments. Peak calcium current densities at +10 mV were as follows: P0, 3.6 and 5.4 pA/pF; P6, 10.9 and 9.9 pA/pF.
Figure 6 compares normalized peak sodium currents (Vtest = −10 mV) in motoneurons frommedtg mice with those from their heterozygous and homozygous normal littermates. The sodium currents from heterozygous motoneurons were not significantly different (p> 0.05) from those of normal motoneurons at any of the three age groups examined (P0–P1, P2–P3, and P7–P8). Moreover, sodium currents in medtg motoneurons were not significantly different from those of heterozygous or homozygous cells at P0–P1 (p > 0.05). However, average peak sodium current density in medtg motoneurons was slightly reduced at P2–P3 and substantially diminished at P7–P8 (for both age groups, p < 0.05 for medtg cells compared with either heterozygous or homozygous cells). Especially at P7–P8, when the yield of viable cells from the dissociation procedure was ∼10-fold lower than in younger animals, recordings were made not only from DiI-labeled cells but also from unlabeled cells identified morphologically as motoneurons (Fig. 6, circles andtriangles, respectively). For none of the three genotypes of P7–P8 cells was there a significant difference (p > 0.05) in sodium current density between labeled and unlabeled cells.

Peak sodium current density as a function of postnatal age in motoneurons from P0–P1, P2–P3, and P7–P8 animals that were wild-type (shaded symbols), heterozygous (filled symbols), or medtg(open symbols). The numbers of cells for the three age groups were 4, 7, and 11 (wild-type), 4, 7, and 10 (heterozygous), and 6, 9, and 11 (medtg), respectively. Cells identified as motoneurons by DiI labeling are indicated withcircles, and cells not identified by DiI labeling are indicated by triangles.
DISCUSSION
We have found that sodium current density increases severalfold in motoneurons isolated from normal mice of increasing postnatal age (P0–P8). We also found that the transcript for the Scn8asodium channel is present in the spinal cord of normal neonatal mice. In motoneurons from medtg mice, which lack a functional Scn8a gene, we did not observe the postnatal increase in average sodium current density. These results are compatible with the hypothesis that postnatal upregulation ofScn8a expression is responsible for much of the increase in sodium current density in normal motoneurons. As previously observed for cerebellar Purkinje cells (Raman et al., 1997), the current density in motoneurons from heterozygous null mice did not differ from the normal wild-type level (Fig. 6). Thus, the loss of one functional allele does not compromise the number of channels in the membrane.
Because acutely isolated motoneurons have fairly extensive processes and are thus poorly suited for voltage-clamp measurements, we used motoneurons isolated and cultured overnight. Cultured motoneuronal somata are likely to express ion channels that would have been targeted to other cellular structures (dendrites, axon, and axon terminal)in vivo. This has been shown to be the case for cultured neurons from the squid stellate ganglion; sodium channels, which are only found in the axon in vivo, appear in the cell bodies after the neurons have been axotomized and placed in culture (Gilly and Brismar, 1989). Our measurements on cultured motoneurons do not allow us to say which cellular structure(s) would have been the destination for the sodium channels that produced the observed postnatal increase in current density. An obvious possibility is that these sodium channels would have been targeted to the axon. Consistent with this possibility, the time frame of sodium current upregulation found in the present study corresponds to that reported previously for increased numbers of sodium channels at nodes of Ranvier in rat peripheral nerve (Vabnick et al., 1996).
Our knowledge remains quite fragmentary as to the identity and subcellular distribution of sodium channel isoforms in motoneurons. Based on in situ hybridization, the Scn8a mRNA is expressed at high levels in adult rat motoneurons (Schaller et al., 1995), and an RNase protection assay indicates that the transcript is also present in the spinal cord of P0 and P7 mice (Fig. 3). Thus, it seems reasonable to attribute the postnatal increase we have observed in motoneuronal sodium current density to an increase inScn8a transcript within motoneurons, particularly because the increase does not occur in animals lacking a functionalScn8a gene. However, it is also possible thatScn8a activity influences the expression of other sodium channel genes in motoneurons.
Northern blot analysis has revealed that the rat spinal cord also contains mRNAs for the RI, RII, and RIII sodium channels, with levels of RII and RIII being relatively high at birth and subsequently declining, and levels of RI being low at birth and increasing postnatally (Beckh et al., 1989). If motoneurons follow the same general pattern as the spinal cord as a whole, then RII and/or RIII channels might account for much of the sodium current present in P0 motoneurons of both normal and medtg mice. Moreover, RI channels have been shown by immunohistochemistry to be present in adult rat motoneurons (Westenbroek et al., 1989) and may contribute to the postnatal increase of sodium current density described here.
It is not entirely clear why the absence of Scn8a sodium channels causes both the cerebellar abnormalities and progressive neuromuscular weakness characteristic of motor end plate disease (Duchen et al., 1967; Duchen, 1970; Duchen and Stefani, 1971). That both cerebellum and motoneurons are affected is consistent with the evidence from in situ hybridization that indicates thatScn8a transcript is widely distributed in the CNS, including motoneurons and cerebellar granule and Purkinje cells (Schaller et al., 1995; Felts et al., 1997). Interestingly, the neuromuscular weakness in mice lacking Scn8a is more severe in the proximal musculature (Duchen, 1970), and the extent of neuromuscular failure varies considerably from muscle to muscle within a limb (Duchen and Stefani, 1971; Harris and Pollard, 1986). Thus, the dysfunction in these mice shows heterogeneity within the motoneuron population. Consistent with such heterogeneity, we found that some motoneurons frommedtg mice had a sodium current density that was only slightly subnormal, whereas most had small currents.
Neuromuscular failure in motor end plate disease may be explained ifScn8a is required for propagation of action potentials along motor axons or for excitation–secretion coupling at motor nerve terminals. Duchen and Stefani (1971) demonstrated that stimulating the biceps nerve in affected mice elicited little or no twitch, although the response to direct electrical stimulation of the muscle was normal. Using microelectrode penetrations of the endoneurium, Duchen and Stefani (1971) were able to record extracellular action potentials from intramuscular branches of the nerve and thus concluded that peripheral nerves remained capable of generating and propagating action potentials, at least down to the intramuscular branches. Intracellular recordings from end plate regions of paralyzed muscles have revealed that raising the extracellular potassium from 5 to 20 mmcauses an acceleration of spontaneous neurotransmitter release in mutant mice similar to that in neuromuscular preparations from normal mice (Duchen and Stefani, 1971; Harris and Pollard, 1985). This suggests that the coupling between depolarization and transmitter release is normal, consistent with our observation of normal calcium currents in medtg motoneurons.
Because muscle nerves contain sensory fibers in addition to motor axons (cf. Boyd and Davey, 1968), our observation of reduced sodium currents in medtg motoneurons may be reconcilable with the observation of Duchen and Stefani (1971) that propagated action potentials are detected by subendoneurial recordings. Specifically, it may be that these action potentials were antidromically propagated via sensory fibers from the site of stimulation (the cut end of the biceps nerve) to the site of recording (intramuscular branches of the nerve). Another possible way to reconcile our results and those of Duchen and Stefani is to postulate that in motor end plate disease, action potential generation in motor nerves is compromised, but only at sites distal to the major intramuscular branches. In this regard, it is interesting that extracellular recordings reveal the presence of sodium channels in the unmyelinated terminals of motor axons (Mallart and Brigant, 1982; Konishi, 1985). Immunohistochemical studies to determine whether Scn8a sodium channels are present in nodes of Ranvier and/or presynaptic terminals will be invaluable for understanding the function of these channels in normal motoneurons and the reason that their absence produces motor end plate disease.
Footnotes
This research was supported by National Institutes of Health Grants NS34509 (M.H.M.), HL02972 (L.K.S.), and NS24444 (K.G.B.) and a Muscular Dystrophy Association grant to Jesús Garcı́a. We thank Jesús Garcı́a for assistance in performing some of the measurements of sodium currents.
Correspondence should be addressed to Kurt Beam, Department of Anatomy and Neurobiology, Colorado State University, Fort Collins, CO 80523-1670.
Dr. Garcı́a’s present address: Department of Physiology and Biophysics, University of Illinois at Chicago, 900 South Ashland Avenue, Chicago, IL 60607.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}