Article Text

Abstract
Background and aims: The aim of this study was to investigate the genetic aetiology of intrahepatic cholestasis of pregnancy (ICP) and the impact of known cholestasis genes (BSEP, FIC1, and MDR3) on the development of this disease.
Patients and methods: Sixty nine Finnish ICP patients were prospectively interviewed for a family history of ICP, and clinical features were compared in patients with familial ICP (patients with a positive family history, n=11) and sporadic patients (patients with no known family history of ICP, n=58). For molecular genetic analysis, 16 individuals from two independently ascertained Finnish ICP families were genotyped for the flanking markers for BSEP, FIC1, and MDR3.
Results: The pedigree structures in 16% (11/69) of patients suggested dominant inheritance. Patients with familial ICP had higher serum aminotransferase levels and a higher recurrence risk (92% v 40%). Both segregation of haplotypes and multipoint linkage analysis excluded BSEP, FIC1, and MDR3 genes in the studied pedigrees. Additionally, the MDR3 gene, previously shown to harbour mutations in ICP patients, was negative for mutations when sequenced in four affected individuals from the two families.
Conclusions: These results support the hypothesis that the aetiology of ICP is heterogeneous and that ICP is due to a genetic predisposition in a proportion of patients. The results of molecular genetic analysis further suggest that the previously identified three cholestasis genes are not likely to be implicated in these Finnish ICP families with dominant inheritance.
- intrahepatic cholestasis of pregnancy
- obstetric cholestasis
- linkage analysis
- ICP, intrahepatic cholestasis of pregnancy
- BSEP, bile salt export pump gene
- FIC1, familial intrahepatic cholestasis 1 gene
- MDR3, multiple drug resistance 3 gene
- PFIC, progressive familial intrahepatic cholestasis
- ATP8B1, ATPase class I type 8B
- BRIC, benign recurrent intrahepatic cholestasis
- ABCB11, ATP binding cassette subfamily B member 11
- γ-GT, gammaglutamyl transferase
- PGY3, P-glycoprotein 3
- HBV, hepatitis B virus
- HCV, hepatitis C virus
- ASAT, serum aspartate aminotransferase
- ALAT, serum alanine aminotransferase
- LOD score, logarithm of odds score
- PCR, polymerase chain reaction
- dNTPs, deoxynucleotidetriphosphates
Statistics from Altmetric.com
- ICP, intrahepatic cholestasis of pregnancy
- BSEP, bile salt export pump gene
- FIC1, familial intrahepatic cholestasis 1 gene
- MDR3, multiple drug resistance 3 gene
- PFIC, progressive familial intrahepatic cholestasis
- ATP8B1, ATPase class I type 8B
- BRIC, benign recurrent intrahepatic cholestasis
- ABCB11, ATP binding cassette subfamily B member 11
- γ-GT, gammaglutamyl transferase
- PGY3, P-glycoprotein 3
- HBV, hepatitis B virus
- HCV, hepatitis C virus
- ASAT, serum aspartate aminotransferase
- ALAT, serum alanine aminotransferase
- LOD score, logarithm of odds score
- PCR, polymerase chain reaction
- dNTPs, deoxynucleotidetriphosphates
Intrahepatic cholestasis of pregnancy (ICP) is a cholestatic disorder which complicates otherwise normal pregnancies. The diagnosis is based on elevated serum levels of bile acid and aminotransferases, and on the presence of pruritus. ICP causes no risk to the mother but it increases the risk of preterm delivery and fetal loss.1 In 0.5–3.5% of ICP cases, the fetus dies suddenly for unknown reasons.2–4
The aetiology of ICP is not known but MDR3 mutations have recently been identified in two families: one with progressive familial intrahepatic cholestasis (PFIC) and ICP5 and another with a single patient with ICP.6 Obligate carriers of PFIC1 and PFIC2 have also been reported to have experienced ICP.7–9 Familial clustering and uneven geographical distribution also indicate the genetic basis of the disease. In Scandinavian countries, ICP occurs in 0.5–1.8% of all pregnancies,10,11 in Canada and Western Europe in 0.1–0.2%,12 while in Chile the reported incidence in pregnancies of women of Araucanian Indian origin is as high as 22%.13 The direct parent to child transmission of ICP suggests dominant inheritance, either autosomal or X linked.14–16
The prevalence of cholelithiasis is known to be higher in ICP patients than in the normal population.17,18 This finding was further strengthened by the recent identification of MDR3 mutations in patients with cholesterol cholelithiasis.19
While the aetiology of ICP is not known, other inherited cholestatic conditions are caused by known gene defects.20–22 Progressive familial intrahepatic cholestasis type 1 (PFIC1) is an autosomal recessive disease characterised by defective biliary secretion of bile acids and was first described in patients of Amish ancestry.7 It starts in infancy and necessitates liver transplantation in the first decade of life. The FIC1 gene (ATPase class I type 8B (ATP8B1)) underlying the disease is located on chromosome 18q21.8 The protein encoded by this gene probably functions as a canalicular P type ATPase that participates in maintaining the distribution of aminophospholipids between the inner and outer leaflets of the plasma membrane.23 Recurrent familial intrahepatic cholestasis (benign recurrent intrahepatic cholestasis (BRIC)) is characterised by recurrent attacks of cholestasis but without progression to chronic liver disease. Consistent with PFIC1, gammaglutamyl transferase (γ- GT) is not increased. The gene involved in this disease was also mapped to chromosome 18q2124 and the gene (ATP8B1) causing PFIC1 disease was subsequently shown to be mutated in BRIC patients.22 A canalicular bile acid transport pump is defective in patients with PFIC type 2 (PFIC2).25 The BSEP (bile salt export pump) gene, also called ATP binding cassette subfamily B member 11 (ABCB11), underlying this disease is located on chromosome 2q24. It encodes a canalicular bile salt export pump, a P-glycoprotein belonging to class B of the ABC transporter super family.
PFIC type 3 (PFIC3) is characterised by defective biliary secretion of phospholipids. Bile becomes toxic because of the lack of phospholipids and therefore causes liver damage. The gene underlying this disease is MDR3 (or P-glycoprotein 3, PGY3) on chromosome 7q21.26,27 Serum γ-GT activity, a differential marker for a MDR3 defect versus FIC1 or BSEP defects, is markedly elevated in these patients. Although symptoms present later in life in PFIC3 than in PFIC1 and PFIC2, liver transplantation is eventually needed. Two reported families with PFIC3 also have ICP.5 Children with PFIC3 are homozygous for mutations in the MDR3 gene while their heterozygous mothers have ICP. One ICP patient with raised serum γ-GT but without a known family history of PFIC has been reported to carry a trafficking mutation in the MDR3 gene.6
SUBJECTS AND METHODS
Subjects and samples
All women with ICP attending the Department of Obstetrics and Gynecology, Helsinki University Central Hospital, between 5 March 1992 and 14 April 1993, and fulfilling the study criteria for ICP (serum aspartate aminotransferase (ASAT) >35 U/l, serum alanine aminotransferase (ALAT) >35 U/l, and/or serum bile acid values >8 mol/l with itching, or serum bile acid values >8 mol/l with or without itching and negative for hepatitis B (HBV) and C (HCV) virus using HBV surface antigen (HBsAg) and HCV antibody testing) were prospectively interviewed for occurrence of ICP and cholelithiasis in first, second, and third degree relatives. The diagnoses of the reported cases of ICP were confirmed from the hospital records. Data on occurrence of cholelithiasis or other liver diseases were recorded according to information provided by the patients. Also, data on previous use of the contraceptive pill were collected.
Each ongoing pregnancy was evaluated for clinical symptoms and laboratory tests and the two highest ASAT, ALAT, bile acid, and bilirubin values during pregnancy and after delivery were recorded. Upper abdomen ultrasound examination was performed to determine if the patient had gall stones. In addition, all relevant clinical variables such as gestational age, birth weight, mode of delivery, and perinatal morbidity were registered for each pregnancy.
Patients with a verified family history of ICP were classified as familial ICP (fig 1) while those without a family history (or for whom the family history could not be verified) were termed sporadic cases. Data were analysed in these two groups of patients. Probability values were calculated using the χ2 and Student’s t test for enumeration and measurement data, respectively. Yates’ correction was used where there was one degree of freedom in the χ2 test. Values of p<0.05 were considered significant.

Eleven pedigrees with familial occurrence of intrahepatic cholestasis of pregnancy.
Two multiplex families with ICP were independently ascertained (pedigrees are shown in fig 2). These included 15 affected and six unaffected individuals from whom venous blood (20 ml) was collected after informed consent. DNA was extracted using standard methods.

{kind=link}
{kind=link}
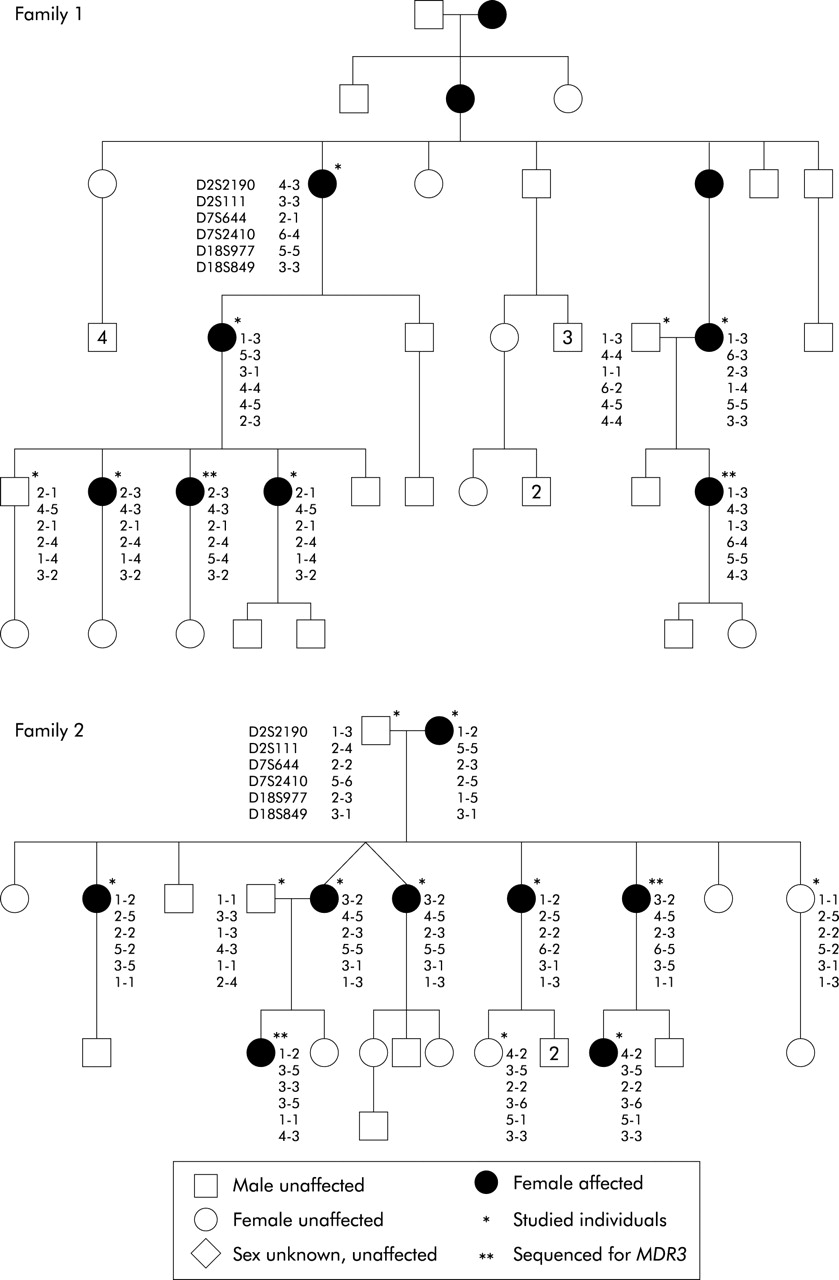
Two Finnish families with intrahepatic cholestasis of pregnancy in which linkage studies were performed. The alleles for the markers D2S2190, D2S111, D7S644, D7S2410, D18S977, and D18S849 are shown for each genotyped individual.
Genotyping
Each individual from the two families depicted in fig 2 was genotyped for the DNA microsatellite markers D2S2190 and D2S111 flanking the BSEP locus, D7S644 and D7S2410 flanking the MDR3 locus, and D18S977 (an intragenic marker) and D18S849 flanking the FIC1 locus. The markers were chosen from the GDB database including Marshfield and Genethon markers. In all cases the forward primer was modified at the 5′ end with a FAM, TET, or HEX fluorescent label.
Polymerase chain reactions (PCR) were performed under the following conditions: 50 ng genomic DNA, 1×buffer (Applied Biosystems, Roche, Branchburg, New Jersey, USA), 10 mM deoxynucleotidetriphosphates (dNTPs), 5 pmol of both the forward and reverse primer, and 0.75 U AmpliTaqGold polymerase (Applied Biosystems) in a final volume of 15 μl. The initial denaturation at 94°C for 10 minutes was followed by 30 cycles (FAM and TET markers) or 35 cycles (HEX markers) at 94°C for 30 seconds, 55°C for 75 seconds, and 72°C for one minute, followed by a final elongation step at 72°C for 10 minutes. Amplified products were separated by electrophoresis on a 4.25% polyacrylamide-6 M urea gel by a 377 DNA sequencer apparatus (Applied Biosystems) and the results were processed by GENESCAN version 2.0.2 and GENOTYPER version 1.1 software.
Linkage and haplotype analysis
Using the simulation program SLINK,28 families 1 and 2 (fig 2) were estimated to give a maximum two point logarithm of odds (LOD) score of 1.47 and 1.99, respectively (recombination fraction [θ]=0.00, marker heterozygosity=0.7, autosomal dominant model with penetrance=85% and phenocopy rate=1.1%). We used the GENEHUNTER 2 program27 to calculate multipoint LOD scores across the BSEP, FIC1, and MDR3 regions, using the same assumptions as in the simulation study and a population frequency of 0.002 for the disease allele assumed on the basis of our own results. The allele frequencies of the polymorphic markers were assumed to be equal.
MDR3 gene sequencing
For mutational analysis of the MDR3 gene, four patients (two individuals from each of the pedigrees in fig 2) were analysed together with a normal control individual. Twenty seven pairs of exon specific primers had been designed for a previous study using long range PCR products covering parts of the MDR3 gene and published sequence information.6 The primers were utilised to amplify the 27 coding exons of the MDR3 gene together with the respective exon/intron boundaries. Amplification of genomic DNA by PCR was carried out using an automated DNA thermal cycler (MJ Research Tetrad, Genetics Research Instrumentation Ltd, Felstead, Essex, UK) in a total volume of 50 μl containing 50–100 ng DNA template, 100 ng of each oligonucleotide primer, 50 mM KCl, 10 mM Tris/HCl, pH 8.3, 1–3 mM MgCl2, 0.01% gelatin, 100 mM dNTP, and 2 units Taq polymerase (Bioline, London, UK). Reaction conditions were as follows: 96°C for three minutes, 35 cycles of 30 seconds at 94°C, 30 seconds at 55–62°C, and one minute at 72°C, followed by a final extension for 10 minutes at 72°C. Following amplification, products were gel purified and sequenced using the MDR3 specific primers and the FS+ Dye Terminator sequencing kit (PE Applied Biosystems) according to the manufacturer’s instructions. Products were resolved and the sequence determined using a 373XL semi automated sequencer (PE Applied Biosystems), and the sequence analysed utilising the Sequence Analysis, Sequence Navigator, and AutoAssembler software packages (PE Applied Biosystems).
RESULTS
In total, 69 pregnant women with ICP were studied. During the study period there were 5304 deliveries in the study hospital, giving an ICP incidence of 1.3 % (69/5304). The incidence of multiple pregnancies was 2.8% (149/5304) in all deliveries and 36% (25/69) in patients with ICP. The incidence of ICP was 14% (19/137) in twin pregnancies and 43% (6/14) in triplets.
The average age of ICP patients when interviewed was 31 years 9 months. Of these, 46% (32/69) were first pregnancies while 54% (37/69) of patients had one or more previous deliveries. A positive family history of ICP was verified in 16% of patients (11/69) (fig 1) while 84% (58/69) of patients did not have relatives diagnosed with ICP, did not have any knowledge of it, or the information given by the patient could not be confirmed. The incidence of ICP among sisters of patients was 12% (6/49). Of 69 patients, 38 (55%) reported a positive family history for cholelithiasis. There was no significant family history of other liver diseases. In total, 57 of 69 patients had used the contraceptive pill. Of these, 6/57 (11%) reported elevated transaminase levels.
In clinical evaluations, 87% (60/69) of patients had itching. In 98% of these cases, symptoms appeared during the third trimester, after the 27th gestational week. Most patients (84%, 57/68) had both elevated aminotransferase and bile acid values. Eight per cent of those who had bilirubin values measured (4/51) were hyperbilirubinaemic (>18 μmol/l) while none of the patients was icteric.
While both serum ASAT (p<0.001) and ALAT (p<0.014) values were higher in the familial than in the sporadic group (table 1), bile acid values showed no significant difference between the two groups. Cholelithiasis was diagnosed with ultrasound, or cholecystectomy had been performed in 22% (14/65) of cases. The percentage of patients with cholelithiasis or who had undergone cholecystectomy was 30% (3/10) in the familial and 20% (11/55) in the sporadic group. The overall recurrence rate of ICP was 51% (29/57). There was a statistically significant difference between the occurrence of ICP in previous pregnancies of familial (92%, 11/12) and sporadic (40%, 18/45) patients. The corresponding values for singleton pregnancies only were 100% (9/9) and 58% (19/33), respectively. Of the multiple pregnancies, 88% (22/25) occurred in the sporadic group.
Two highest ASAT, ALAT, and bile acid values, and comparison of clinical features of ICP between familial and sporadic cases
The pedigrees and haplotypes of the two families in which linkage analysis was performed are shown in fig 2. The distance between the flanking markers D2S2190 and D2S111 at the BSEP locus on chromosome 2q24 is 4.8 cM. Multipoint linkage analysis yielded positive LOD scores over this region in family 2, with a maximum 1.12, while the results were negative in family 1. In haplotype analysis, the affected individuals in family 1 had inherited different alleles, further excluding BSEP in family 1.
The distance between the markers flanking the MDR3 gene on chromosome 7q21 (D7S644, D7S2410) is 2.9 cM. The total LOD scores were negative across the interval (table 2). As MDR3 has previously been implicated in ICP,5,6 we sequenced two patients from both families (see fig 2) but no mutations were identified.
Multipoint linkage analysis in two Finnish ICP pedigrees
The distance between the flanking markers D18S977 and D18S849 in the FIC1 region on chromosome 18q21 is 1.5 cM. Multipoint linkage analysis in this interval yielded clearly negative LOD scores. In both families affected family members had differing haplotypes.
DISCUSSION
The frequency of ICP varies widely in different populations. We found ICP in 1.3% of pregnancies which coincidences with previous reports from Finland10,16 It is possible that this is an underestimation of the actual incidence of ICP if all cases are not diagnosed. However, in Finland this would be unlikely as serum aminotransferase levels are routinely measured if a patient has itching, in multiple pregnancies, and in patients with a history of ICP or itching in a previous pregnancy. If aminotransferase levels are elevated, the patient is referred to the same maternity hospital (according to the residential address) in which the patient would deliver in the case of an uneventful normal pregnancy. This study was performed in one of these hospitals in the Helsinki area.
The high incidence of multiple pregnancies (36%) and increasing incidence of ICP in twin (14%) and triplet (43%) pregnancies support the current view of multiple pregnancy being a risk factor for ICP. In fact, this may be the only predisposing factor in some patients while ICP may be more likely to occur in singleton pregnancies in patients with an inherited predisposition.
ICP occurred in at least two close relatives in 11 of 69 (16%) families. Only those in whom the family history could be verified from hospital records were accepted as familial. However, it is likely that some familial cases were missed, as it was not possible to confirm the diagnosis of ICP in pregnancies prior to the late 1960s. Therefore, 16% is the minimum proportion of familial cases and this number may actually be higher.
Several differences in clinical features were noted between familial and sporadic cases. Familial cases had significantly higher serum transaminase levels and, in particular, a higher frequency of ICP (92% v 40%) in previous pregnancies. These differences in phenotype may reflect a difference in the underlying aetiology of ICP in the two groups. However, as there are no definite tests to identify familial ICP, one would expect some of the familial cases to be identified as sporadic—that is, due to inheritance of the risk allele through males in previous generations. Therefore, it may not always be possible to show clearcut differences between the two groups, even if they existed.
The prevalence of cholelithiasis was 22% in ICP patients. While there are no data on the prevalence of cholelithiasis in healthy women of reproductive age in Finland, studies from Denmark and Norway reported a prevalence of 4.8–15.3%.30,31 Thus the higher frequency among ICP patients in this study also suggests an association between ICP and cholelithiasis.14–16 Interestingly, 55% of patients had relatives who had been diagnosed with cholelithiasis or were cholecystectomised. Additionally, several male relatives of index patients were affected with cholelithiasis and cholelithiasis was more frequent in the familial than in the sporadic group. Therefore, cholelithiasis may be another expression of liver dysfunction causing ICP, as suggested by the recent finding of MDR3 mutations in patients with ICP and in patients with cholesterol cholelithiasis.19
The dominant mode of inheritance of ICP, either autosomal or X linked, has been proposed.14–16 The lacking male phenotype, incomplete penetrance, and existence of ICP only in pregnant women all complicate the study of inheritance of ICP. Of the 11 familial cases, pedigrees 1, 2, and 3 showed typical dominant inheritance. In the other eight pedigrees, dominant inheritance was possible considering the unknown male phenotype and incomplete penetrance although they did not clearly display the pattern. The two large pedigrees in which linkage studies were performed both showed clear dominant inheritance (fig 2). However, it is not possible to exclude X linked inheritance by showing male to male transmission in a female limited disease.
Linkage and haplotype analysis of BSEP, FIC1, and MDR3 loci did not indicate that these genes were implicated in the Finnish ICP families. In family 1, linkage analysis of the flanking markers around the BSEP gene yielded negative LOD scores and inheritance of different haplotypes from the affected mother to her affected descendants further excluded the involvement of the BSEP gene in this family, although slightly positive LOD scores were found in family 2. The FIC1 gene was clearly excluded by both haplotype and linkage analysis in Finnish ICP patients. The negative total LOD scores indicate exclusion of the MDR3 gene, as do the results of sequence analysis which were negative for mutations when four affected individuals from the two families were studied.
In conclusion, the aetiology of ICP appears to be heterogeneous. At least in 16% of cases ICP is familial and therefore likely to be caused by an inherited predisposition, which is dominantly inherited. The present data suggest that MDR3 and two other genes (FICI, VSEP) causing cholestatic conditions are not likely to be involved in Finnish ICP families and that other unknown genes may underlie ICP.
Acknowledgments
We are greatly indebted both to Ms Sinikka Lindh for keeping in contact with the patients, collecting the blood samples, and for her help in many other ways, and to Dr Kari Teramo for his invaluable expert opinions in obstetrics when this study was started. This study was funded by Sigrid Juselius Foundation and Finnish State grants TYH 8320 and TYH 2210.
REFERENCES
Footnotes
-
↵* M Savander, K Avela, and A Ropponen contributed equally to this work