Article Text

Abstract
Objective Hodgkin's lymphoma survivors who were treated with infradiaphragmatic radiotherapy or procarbazine-containing chemotherapy have a fivefold increased risk of developing colorectal cancer (CRC). This study aims to provide insight into the development of therapy-related CRC (t-CRC) by evaluating histopathological and molecular characteristics.
Design 54 t-CRCs diagnosed in a Hodgkin's lymphoma survivor cohort were analysed for mismatch repair (MMR) proteins by immunohistochemistry, microsatellite instability (MSI) and KRAS/BRAF mutations. MSI t-CRCs were evaluated for promoter methylation and mutations in MMR genes. Pathogenicity of MMR gene mutations was evaluated by in silico predictions and functional analyses. Frequencies were compared with a general population cohort of CRC (n=1111).
Results KRAS and BRAF mutations were present in 41% and 15% t-CRCs, respectively. Compared with CRCs in the general population, t-CRCs had a higher MSI frequency (24% vs 11%, p=0.003) and more frequent loss of MSH2/MSH6 staining (13% vs 1%, p<0.001). Loss of MLH1/PMS2 staining and MLH1 promoter methylation were equally common in t-CRCs and the general population. In MSI CRCs without MLH1 promoter methylation, double somatic MMR gene mutations (or loss of heterozygosity as second hit) were detected in 7/10 (70%) t-CRCs and 8/36 (22%) CRCs in the general population (p=0.008). These MMR gene mutations in t-CRCs were classified as pathogenic. MSI t-CRC cases could not be ascribed to Lynch syndrome.
Conclusions We have demonstrated a higher frequency of MSI among t-CRCs, which results from somatic MMR gene mutations. This suggests a novel association of somatic MMR gene mutations with prior anticancer treatment.
- COLORECTAL CANCER
- MICROSATELLITE INSTABILITY
- RADIATION THERAPY
- CHEMOTHERAPY
- DNA DAMAGE
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Hodgkin's lymphoma survivors have an increased risk of developing colorectal cancer.
This increased colorectal cancer risk is associated with the treatment for Hodgkin's lymphoma, including infradiaphragmatic radiotherapy and procarbazine-containing chemotherapy.
Knowledge on the molecular pathways of therapy-related colorectal cancer is very limited.
What are the new findings?
Therapy-related colorectal cancer is heterogeneous in terms of microsatellite instability (MSI) status, CpG island methylator phenotype status and mutation status of several oncogenes.
Therapy-related colorectal cancer has a higher frequency of MSI compared with colorectal cancer in the general population.
The higher MSI frequency results from somatic mismatch repair gene mutations, which are rare in colorectal cancer in the general population.
This suggests a novel association of somatic mismatch repair gene mutations with prior anticancer treatment.
How might it impact on clinical practice in the foreseeable future?
MSI status should be determined in all colorectal cancer patients with a history of Hodgkin's lymphoma.
When MSI colorectal cancer is diagnosed in patients who previously have been treated with alkylating agents and/or infradiaphragmatic radiotherapy, analysis of both germline and somatic mutations in mismatch repair genes should be considered.
Background
Approximately 18% of all cancers are diagnosed in cancer survivors.1 These second primary malignancies develop due to multiple factors, including genetic predisposition, environmental factors and lifestyle factors.2 In addition, certain cancer treatments have the capability to cause cancer, because of their mutagenic and genome destabilising effects also affect normal cells.
After treatment of various primary malignancies, the risk of developing colorectal cancer (CRC) increases. This increased risk ranges from a 2-fold to 11-fold compared with an age-matched control population and this effect persists for decades.3–8 Increased risks of CRC have been reported in survivors of Hodgkin's lymphoma, testicular cancer, Wilms tumour, central nervous system malignancies, bone cancer and prostate cancer, among others.
In Hodgkin's lymphoma survivors, second solid malignancies are a major cause of morbidity and mortality. After breast cancer and lung cancer, CRC is the third most common second solid malignancy in Hodgkin's lymphoma survivors.3 ,8 ,9 Both abdominal irradiation and alkylating agents (procarbazine) have been associated with this increased CRC risk.3 ,5 ,6 ,8 ,10
Knowledge of the mechanisms behind CRC development after radiotherapy (RT) and/or chemotherapy exposure is very limited. A recent review on case reports of RT-related rectal cancer reported a high frequency of chronic radiation colitis as well as mucinous adenocarcinomas, which would possibly be sequentially related.11
The present study aims to provide more insight in the pathogenesis of RT-related and chemotherapy-related CRC (t-CRC) in Hodgkin's lymphoma survivors by histopathological and molecular analyses.
Materials and methods
Patients and tissue samples
A total of 62 patients with t-CRC were identified in a large multicentre cohort of Hodgkin's lymphoma patients (N=3905; treatment period 1965–2000), who survived at least 5 years after diagnosis of Hodgkin's lymphoma. Demographic data and data on Hodgkin's lymphoma treatment, follow-up and second primary malignancies (type, location, histology, stage) were readily available, as they had been collected through historical hospital-based registries or cancer registries as previously described.8 ,12 ,13
Formalin-fixed paraffin-embedded (FFPE) tissue and pathology reports of all t-CRCs in Hodgkin's lymphoma survivors were requested through the nationwide network and registry of histopathology and cytopathology in the Netherlands.14
This study received approval of the translational research board of the Netherlands Cancer Institute (study number CFMPB208). All data and material were processed anonymously. Privacy restrictions prevented us from obtaining additional clinical information. Collection, storage and use of patient-derived tissue and data were performed in compliance with the ‘Code for Proper Secondary Use of Human Tissue in The Netherlands’, Dutch Federation of Biomedical Scientific Societies, the Netherlands.
Histopathology
All H&E stained slides were reassessed according to a standard protocol. Variables collected included tumour location, tumour, node, metastases stage, histological type, grade of differentiation and histological changes in normal colonic tissue indicating radiation damage (eg, vascular and mucosal changes, fibrosis). The presence of distant metastases (M stage) was assessed based on clinical information in pathology reports and tissue samples of metastases when available. In case of synchronous CRCs, both cancers were completely evaluated.
Immunohistochemistry
A tissue microarray was made of all resection specimens. In case of biopsy material, multiple sections for immunohistochemistry (IHC) were cut instead. IHC for mismatch repair (MMR) proteins was performed according to standard protocols for Ventana immunostainer (MLH1 (M1, 6472966001 Roche (Ventana)), MSH2 (G219-1129, 5269270001, Roche (Ventana)), MSH6 (EP49, AC-0047, Epitomics), PMS2 (EP51, M3647, Dako)).
Molecular analyses
DNA from tumour and normal colon or tumour-negative lymph nodes (in the absence of normal colon tissue) was isolated using a Qiagen extraction kit. DNA concentrations were measured using the Qubit 2.0 Fluorometer with the Qubit dsDNA Assay Kit.
A pentaplex PCR-based assay for microsatellite instability (MSI) was carried out using fluorescent labelled primers of five mononucleotide repeat targets (BAT25, BAT26, NR24, NR21, NR27), followed by fragment analysis. MSI was defined as instability in two markers or more. Additional analyses on MSI or MMR deficient tumours are described below and an overview is given in figure 1.

Flow chart of MSI and MMR gene analyses. t-CRC, therapy-related colorectal cancer; MSI, microsatellite instable; MMR, mismatch repair; MSS, microsatellite stable.
Promoter methylation of MMR genes was evaluated in MSI tumours by a multiplex ligation-dependent probe amplification (MLPA) kit (ME011-B2 kit; MRC Holland, Amsterdam, the Netherlands). This probemix included a total of 25 probes for the promoter region of six different MMR genes (MLH1, MSH2, MSH6, PMS2, MSH3, MLH3). Gene positivity was defined as 33% of probes per gene with a cut-off for positivity of 0.2 at probe level.
To detect a CpG island methylator phenotype (CIMP) phenotype, a MLPA kit was used to evaluate the methylation status of promoter regions of eight different genes (CACNA1G, CDKN2A, CRABP1, IGF2, MLH1, NEUROG1, RUNX3, SOCS1) with 31 methylation-specific MLPA probes (ME042-B2 kit, MRC Holland). Two scoring alternatives were used.15 The mild scoring method was the Ogino 5/8 gene positivity at a positive gene level of 33% of probes per gene with a cut-off for positivity of 0.2 at probe level. The strict scoring method was the Ogino 6/8 gene positivity at a gene level of 66% of probes with a probe cut-off for positivity of 0.2.16 Non-discriminating probes (positive in 0% or in ≥85% of CRCs) were excluded from the scoring.
Mutations in common cancer-related genes were evaluated using a custom panel of the Molecular Diagnostics Department of the Netherlands Cancer Institute (developed for the Sequenom MassAnalyser, Agena Bioscience, San Diego, California, USA). The panel detects mutations at hotspots of eight oncogenes (AKT1, BRAF, DDR2, EGFR, MEK1, PIK3CA, KRAS, NRAS; see online supplementary table S1).
Supplemental material
Targeted next-generation sequencing analysis
In MSI cases without MMR gene promoter methylation, MMR genes were additionally screened for mutations with the Ion Torrent Personal Genome Machine (PGM), with supplier's materials and protocols (Life Technologies, Carlsbad, California, USA). A custom-made primer panel was used, designed using Ion AmpliSeq Designer 3.0. The panel encompasses 306 amplicons covering the genes for MLH1, MSH2, MSH6 and PMS2 as previously described,17 as well as hot spot mutation regions in POLE (codons 286, 297, 411 and 424) and POLD1 (codon 478). In addition, primer panels for analysis of polymorphic single nucleotide polymorphisms (SNPs) were added for detection of large genomic aberrations or loss of heterozygosity (LOH) of chromosomes 2, 3 and 7. Further details on the panel and the assessed chromosomal regions are presented in online supplementary table S2 and supplementary methods.18 Diagnostic use of SNP-based LOH analysis will be described in detail elsewhere.19
Variants detected by next-generation sequencing analysis were tested in both neoplastic and normal tissue (normal colorectal wall or lymph nodes, as blood was not available) by Sanger sequencing.
Mutations were confirmed by Sanger sequencing and/or a repeated PGM procedure with newly isolated DNA from the same tissue blocks after manual microdissection.17 In MSI t-CRCs without somatic MMR gene mutations, the presence of exon deletions and duplications in specific regions of MLH1 and MSH2 was evaluated by copy number MLPA (P003-B1 kit and P248-B1 confirmation kit, MRC Holland). A tumour versus control ratio of <0.75 was classified as a deletion and >1.30 as a duplication.
The pathogenicity of the detected MMR gene variants was predicted using InSIGHT classification,20 ,21 Align-GVGD,22 SIFT,23 Mutation Taster,24 ,25 PolyPhen-226 and a literature search. The variants were classified as benign (1), likely benign (2), uncertain (3), likely pathogenic (4) or definitely pathogenic (5). In combination with the functional analysis results, the pathogenicity of the variants was interpreted.
Functional analysis of MMR mutations
The functionality of MMR gene variants was tested using the previously described protocol.27 Briefly, cultures of mouse embryonic stem cells (hemizygous for the MMR gene under study) were exposed to single-stranded oligo-DNAs designed to introduce the mutation of interest into the endogenous MMR gene in a subset of cells (see online supplementary table S3 and Houlleberghs et al27). Subsequent exposure of the cell culture to 6-thioguanine (6TG) selects for cells that had become MMR deficient. The presence of the mutation in 6TG-resistant cells (verified by Sanger sequencing) indicates the mutation abrogates MMR activity. This protocol has been developed, validated and used for MSH2 variants27 and extended to MSH6 and MLH1 (Houlleberghs et al, manuscripts in preparation).
Comparison with a cohort of the general population
Frequencies of MSI, MMR IHC and MMR gene mutations were compared with a previously obtained dataset of CRCs in the general population.17 ,28 That study included 1117 patients with CRC diagnosed between 2007 and 2009 at the age of 70 or younger. This young general population cohort was selected because of the availability of the required data (MSI status, MMR status, MLH1 methylation, etc) and the comparability with the t-CRC cohort, as the median age of diagnosis in t-CRC was 57 years (interquartile range (IQR) 50–62). For the current analyses, we excluded six patients from this database, five due to pT0 and one because of being present in our Hodgkin's lymphoma survivor cohort.
Statistical analyses
Descriptive patient characteristics, Hodgkin's lymphoma characteristics and CRC characteristics were analysed using IBM SPSS V.22.0 database software. Frequencies of tumour characteristics were compared with the general population cohort using χ2 test or Fisher's exact test for binary or categorical data and Mann-Whitney U test for continuous data that did not have a normal distribution. Two-sided p≤0.05 was considered as a significant difference.
Results
Characteristics of Hodgkin's lymphoma survivors who developed CRC
FFPE tissue and pathology reports were obtained for 51/62 (82%) patients with t-CRC diagnosed in Hodgkin's lymphoma survivors (see online supplementary table S4). Hodgkin's lymphoma was diagnosed at a median age of 31 years (range 15–49 years, table 1). In 44/51 (86%) of patients, Hodgkin's lymphoma was treated with agents that have been associated with an increased CRC risk, that is, infradiaphragmatic RT and/or procarbazine.3 ,8 A subset of patients (6/51, 12%) received neither of these treatments, but did receive supradiaphragmatic RT and/or other chemotherapeutics. In one case, treatment details were missing.
HL characteristics
General characteristics of therapy-related CRC
t-CRCs were diagnosed a median period of 22 years after Hodgkin's lymphoma diagnosis (range 7–39 years), between 1988 and 2014 (median 2002). The median age at t-CRC diagnosis was 57 years (range 30–79 years, table 2).
Characteristics of t-CRCs and CRCs in general population
Despite the selection of a young reference population, t-CRCs in the Hodgkin's lymphoma survivor cohort were diagnosed at a younger median age. Three patients were diagnosed with a synchronous t-CRC, which resulted in a total number of 54 t-CRCs. t-CRCs were more frequently present in the proximal colon compared with CRCs in the general population (24/54, 45% vs 27%, p=0.003). This included nine t-CRCs (17%) that were located in the transverse colon compared with 4% of CRCs in the general population (p<0.001). No morphological signs of previous radiation were detected in the resection specimens that could be evaluated (n=31). In 20 cases, this could not be evaluated due to neoadjuvant (chemo)-RT in 5 cases of rectal cancer, and absence of evaluable normal mucosa in 15 cases.
Loss of MSH2 and MSH6 staining is more frequent in therapy-related CRC
The frequency of MSI was higher in t-CRCs than in CRCs in the general population (13/54, 24% vs 11%, p=0.003, table 2). Loss of MSH2 and MSH6 staining was more frequently present in t-CRCs compared with CRCs in the general population (7/54, 13% vs 1%, p<0.001). The frequency of loss of staining of MLH1 and PMS2 was similar in both groups (5/54 (9%) in t-CRCs vs 8% in the general population, p=0.761). One MSI t-CRC had positive nuclear staining of all four MMR proteins. Three out of five MSI t-CRCs with loss of MLH1 and PMS2 staining were explained by MLH1 promoter methylation. The presence of MSI as a consequence of MLH1 promoter methylation was equal in both groups (3/54, 6% vs 6%, p=0.806). However, the frequency of MSI without MLH1 promoter methylation was higher in t-CRCs (10/54, 19% vs 5%, p<0.001).
In the three patients with two synchronous t-CRCs, only one t-CRC was MSI with MLH1 promoter methylation and the other five t-CRCs were microsatellite stable (MSS).
MMR genes were more frequently mutated in MSI therapy-related CRCs
Of the 10 MSI t-CRCs without MLH1 promoter methylation, 7 had a loss of MSH2 and MSH6 staining, 2 had a loss of MLH1 and PMS2 staining and 1 had positive staining of all four MMR proteins. In addition, one MSS t-CRC with an isolated loss of PMS2 staining did not show MLH1 promoter methylation. Because of this high number of unexplained MSI t-CRCs, we additionally performed promoter methylation analysis of multiple MMR genes and mutation analysis of MMR genes, POLE and POLD1 on these 10 MSI t-CRCs and 1 MSS t-CRC with an isolated loss of PMS2 staining (figure 1).
Promoter methylation of MSH2, MSH6, PMS2, MSH3 or MLH3 was absent in these t-CRCs. In addition, POLE and POLD1 mutations were not detected. MMR gene mutations were present in 8/10 MSI t-CRCs and absent in the MSS t-CRC with loss of PMS2 staining (table 3).
MMR gene variants in MSI t-CRCs without MLH1 promoter methylation diagnosed in Hodgkin's lymphoma survivors*
First, the two t-CRCs with loss of MLH1 and PMS2 staining without MLH1 promoter methylation could be explained by pathogenic MLH1 mutations, one tumour containing two pathogenic missense mutations, the other a deleterious splice site mutation and LOH of chromosome 3.
Second, in five out of seven t-CRCs with loss of MSH2 and MSH6 protein staining, pathogenic MMR gene mutations were detected. In two cases, a pathogenic MSH2 mutation was accompanied by LOH of chromosome 2. Remarkably, in the third tumour six de novo mutations were detected: three in MSH2, two in MLH1 and one in MSH6. Two of the MSH2 mutations were pathogenic. The fourth tumour with absent MSH2/MSH6 staining contained a likely pathogenic MSH6 variant, however, with inconclusive LOH results. In the fifth tumour, SNP analysis results clearly demonstrated LOH of the MSH2 locus. In addition, MLPA results indicated a somatic homozygous loss of the MSH2 exon 8 probe region and no aberrations in the other MSH2 exons were observed. These findings point to the presence of MSH2 copy neutral LOH in combination with a homozygous deletion in MSH2 exon 8 in the tumour cells, resulting in biallelic inactivation of the MSH2 gene. The last two out of seven t-CRCs with loss of MSH2/MSH6 staining did not carry a mutation or exon deletion in any MMR gene, but did show LOH of chromosome 2 that harbours MSH2 and MSH6, respectively. This result was confirmed by copy number MLPA in both t-CRCs.
Finally, in the single MSI t-CRC with normal staining of all four MMR proteins, two MSH6 mutations were detected that were classified as pathogenic.
Classification of the MMR gene mutations by in silico predictions and functional analysis were highly concordant (table 3). All the pathogenic mutations were absent in normal tissue and hence somatically acquired. Two t-CRCs had a MMR gene variant that was also present in normal tissue. These variants, MSH2 c.965G>A (p.G322D) and MLH1 c.1207C>T (p.P403S), were classified as non-pathogenic.
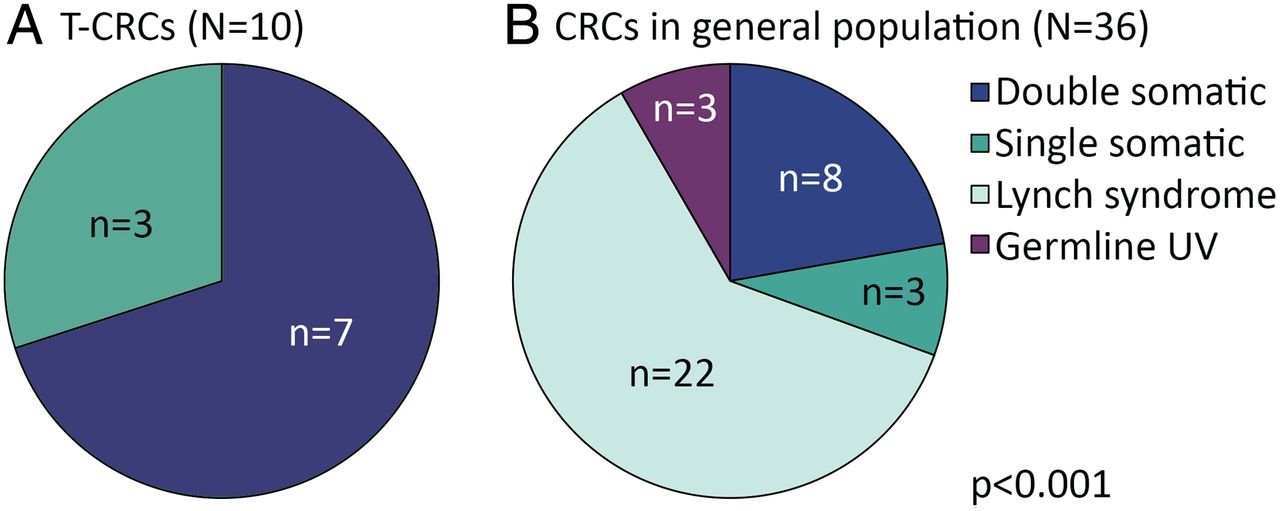
Thus, for 7 out of 10 t-CRCs without MLH1 promoter methylation, we could explain the MSI phenotype of t-CRCs by somatic acquisition of deleterious MMR gene mutations, either affecting both alleles, or accompanied by LOH. The aetiology of MSI in CRCs without MLH1 promoter methylation was different in the t-CRC population and the general population (p<0.001, figure 2). The frequency of somatic MMR gene mutations with a second mutation or LOH (defined as double somatic in figure 2) was higher in MSI t-CRCs without MLH1 promoter methylation (7/10 (70%)) than in MSI CRCs without MLH1 promoter methylation in the general population (8/36 (22%) Fisher's exact test p=0.008). In contrast, Lynch syndrome was the cause for MSI without MLH1 promoter methylation in 22 CRCs in the general population and was not detected in patients with t-CRC.

{kind=link}
{kind=link}
Mismatch repair gene mutations in MSI colorectal cancer without MLH1 methylation. (A) Therapy-related colorectal cancer (t-CRC), (B) 14/50 MSI CRCs without MLH1 promoter methylation in the general population were excluded because no further testing was performed. Fisher’s exact test. UV, unclassified variant; double somatic, two mutations or one mutation and loss of heterozygosity; single somatic, mutation or loss of heterozygosity.
Oncogene mutation status and CIMP were not related to MSI status in therapy-related CRC
A CIMP high phenotype was present in 21 cases (21/53, 39%) using the mild Ogino criteria and in 5 cases (5/53, 9%) using the strict Ogino criteria (table 4).15 ,16 One case was not evaluable for CIMP. KRAS mutations were detected in 22/54 t-CRCs (40%), which included 5 cases with a concurrent PIK3CA mutation (5/54, 9%, table 4). BRAF mutations were present in 8/54 t-CRCs (15%). The panel of eight oncogenes (AKT1, BRAF, DDR2, EGFR, MEK1, PIK3CA, KRAS, NRAS) revealed no mutations in 21/54 CRCs (39%). For both CIMP and oncogene mutation status, no association was found with MSI status.
t-CRC histopathological and molecular characteristics (N=54)
No evidence for a relationship between a specific Hodgkin's lymphoma treatment and t-CRC characteristics
The frequencies of MSI and CIMP did not vary between different Hodgkin's lymphoma treatment groups (abdominal RT alone vs procarbazine alone vs abdominal RT plus procarbazine vs neither, data not shown). Seven out of eight patients with t-CRC with at least one MLH1, MSH2 or MSH6 mutation had previously been treated with abdominal RT (n=2), procarbazine-containing chemotherapy (n=3) or the combination of both treatments (n=2). The patient with two deleterious MSH6 mutations did not receive these treatments, but had received a dacarbazine-containing chemotherapy regimen (an alkylating agent within the triazene family, similar to procarbazine).
Discussion
In this first report on histopathological and molecular characteristics of t-CRC related to Hodgkin's lymphoma treatment, we have demonstrated that similar to sporadic CRC, t-CRCs are heterogeneous in terms of MSI status, CIMP status and mutation status of several oncogenes. Interestingly, we did find a higher frequency of MSI t-CRCs resulting from somatic MMR gene mutations. These somatic mutations are likely related to abdominal RT and/or alkylating agents (eg, procarbazine), as these therapeutics increase the risk of developing CRC.3 ,5 ,6 ,8 ,10
The aetiology of the somatic MMR gene mutations in CRCs has so far not been elucidated. Somatic MMR gene mutations have been described in several recent publications,17 ,29–31 but generally without providing detailed information on the clinical history of these patients, including prior chemotherapy and/or RT.
The main function of the MMR system is recognition and repair of DNA replication errors. These are recognised by the MSH2-MSH6 protein complex, which binds to the mismatch and recruits the MLH1-PMS2 protein complex. Biallelic inactivation of one of the genes encoding the MMR system leads to a disruption of the DNA repair process and consequently to an accumulation of somatic mutations, which subsequently leads to CRCs with MSI and a hypermutator phenotype.32
An interesting link exists between the MMR system, RT and alkylating agents. The MMR system is important in the cellular response to alkylating agents, as the major cytotoxic lesion (O6-methylguanine) leads to a DNA mismatch during replication (O6-methylguanine-thymine) that activates the MMR system.33 Mouse models with inactivation of a MMR gene indeed show an accelerated CRC development after exposure to irradiation or alkylating agents.34–37 In addition, alkylating agents increase the number of MMR-deficient intestinal crypts in mice.34 It can thus be hypothesised that pre-existing epithelial intestinal cells with some level of MMR dysfunction are targeted by RT and/or alkylating agents, which could lead to the development of MSI CRCs.
Chronic radiation colitis has been reported in case series of radiation-related CRC.11 In these reports, a higher frequency of mucinous adenocarcinomas among t-CRCs was suggested, which was not confirmed in our study. In addition, we did not find histological signs for chronic inflammation in our cases. Evidence from mouse models, however, suggests that inflammation also contributes to CRC development after RT or alkylating agents.38 ,39 In a mouse model of chronic inflammation simulating inflammatory bowel disease, a higher frequency of MSI has been reported with a possible relation to base excision repair.40 As base excision repair is also involved in the repair of lesions induced by RT and alkylating agents, this repair mechanism may be interesting to evaluate in future studies of t-CRC.41
Data from other cancer types also provide links between the MMR system and cancer therapy. In glioma patients treated with the alkylating agent temozolomide, the cells that were found at recurrence had reduced MMR protein levels and contained MSH6 gene mutations and a hypermutated profile, which contrasted the primary gliomas before treatment.42–44
The MMR system also plays an important role in therapy-related acute myeloid leukaemia (t-AML) after exposure to alkylating agents or topoisomerase inhibitors. t-AML is classified separately from sporadic AML and has specific clinical and biological characteristics.45–48 t-AML has a high frequency of MSI (40%–90% vs 0%–30% in sporadic AML),49–52 associated with MLH1 promoter methylation or loss of expression of MSH2, possibly caused by deleterious MSH2 mutations.49 ,53 ,54
In other second malignancies, results are conflicting. A higher frequency of MSI has been reported in therapy-related lung cancer.55 In contrast, in therapy-related oesophageal cancer56 and breast cancer,55 the frequency of MSI was not increased, but numbers were very small.
In our study, 3 out of 13 MSI t-CRCs were not explained by either MLH1 promoter methylation or double somatic aberrations of MMR gene mutations. It is possible that mutations have been missed as the MMR genes were not fully sequenced (see online supplementary table S2). Thus, exon mutations outside of the sequenced regions or mutations in intronic regions that may affect gene function could not be detected, and could be somatic or germline. Another possible explanation is that epigenetic alterations or mutations in genes of unknown relevance are present.
Finally, of the 15 de novo acquired MMR gene mutations, 3 did not show up as pathogenic in our in silico and functional tests and 2 were inconclusive. Six mutations were detected in a single tumour that contained only two pathogenic mutations. The appearance of multiple MMR gene mutations without phenotypic consequences in a single tumour has been described previously and remains remarkable and unexplained.17
We acknowledge that our study has a number of limitations. First of all, not all CRCs arising after Hodgkin's lymphoma were necessarily related to the previous therapy. In that regard, information on family history and other risk factors were not available. In addition, our patients with t-CRC were treated for Hodgkin's lymphoma between 1965 and 1999, mainly with abdominal RT and/or procarbazine (44/51, 86%), which increases the risk of developing CRC approximately fivefold.3 ,8 ,9 This means that the attributable risk in this population was 80% ((relative risk–1)/relative risk). Consequently, at least 20% of our cases were probably not related to Hodgkin's lymphoma treatment. Importantly, RT and procarbazine doses in Hodgkin's lymphoma treatment have been reduced over the past decades. Despite these changes, the cumulative incidence of second primary gastrointestinal malignancies did not differ among older and more recently treated patients.8
Finally, our reference general population cohort did not match the t-CRC patient cohort completely with regard to age and stage, which both correlate with MSI frequency. The difference in median age between the two cohorts was, however, minimal, that is, only 4 years. The staging information of the general population cohort was incomplete in 27% of cases, which makes comparison of stages difficult. Still, since MSI is least frequent in stage IV of all stages, it can be argued that the higher frequency of stage IV disease in the t-CRC cohort is likely to strengthen our finding of a higher MSI frequency in t-CRCs compared with the general population.
In conclusion, t-CRCs are genomically heterogeneous and have a higher frequency of MSI compared with CRCs in the general population. This higher MSI frequency is explained by somatic MMR gene mutations. This study is the first to provide a possible aetiology for MSI CRC carrying somatic MMR gene mutations, that is, prior anticancer treatment. When MSI CRC is diagnosed in patients who previously have been treated with alkylating agents and/or RT because of Hodgkin's lymphoma, analysis of both germline and somatic mutations in MMR genes should be considered.
Acknowledgments
We would like to acknowledge the NKI-AVL Core Facility Molecular Pathology & Biobanking (CFMPB) for supplying NKI-AVL Biobank material and/or lab support. We would also like to thank Roelof Pruntel for technical assistance, Anne Goverde for the help with the general population database, Hellen Houlleberghs for help with the functional assay and all participating laboratories for the distribution of the CRC material. Finally, we thank Begoña Diosdado for the thorough review of the manuscript.
References
Footnotes
Contributors Study concept and design: LSR, BMA, GAM, FEvL, MEvL, PS. Acquisition of data: LSR, PS, PNA, TWvR and JH. Analysis and interpretation of data: LSR, PS, WND, EHR, JtH, PNA, WRG-G, GAM, HtR, TWvR, JH, FEvL and MEvL. Drafting of the manuscript: LSR and PS. Critical revision of the manuscript for important intellectual content: all authors. Statistical analysis: LSR, FEvL and MEvL. Obtained funding: MEvL, MLDS funding project FP14-04. Administrative, technical or material support: PALGA group. Study supervision: GAM, HtR, FEvL and MEvL.
Collaborators PALGA collaborators; K Schelfout, HADM van Herk, ID Nagtegaal, JWR Meijer.
Funding Dutch Society of Gastroenterology and Hepatology (Maag Lever Darm Stichting (MLDS) (FP14-04).
Competing interests MEvL obtained funding from the Dutch Society of Gastroenterology and Hepatology (Maag Lever Darm Stichting (MLDS) funding project FP14-04).
Ethics approval Translational Research Board of the Netherlands Cancer Institute (study number CFMPB208).
Provenance and peer review Not commissioned; externally peer reviewed.