Article Text

Abstract
BACKGROUND AND AIMS In familial adenomatous polyposis (FAP), correlations between site of mutation in the adenomatous polyposis coli (APC) gene and severity of colonic polyposis or extracolonic manifestations are well known. While mutation analysis is important for predictive diagnosis in persons at risk, its relevance for clinical management of individual patients is open to question.
METHODS We examined 680 unrelated FAP families for germline mutations in theAPC gene. Clinical information was obtained from 1256 patients.
RESULTS APC mutations were detected in 48% (327/680) of families. Age at diagnosis of FAP based on bowel symptoms and age at diagnosis of colorectal cancer in untreated patients were used as indicators of the severity of the natural course of the disease. A germline mutation was detected in 230 of 404 patients who were diagnosed after onset of bowel symptoms (rectal bleeding, abdominal pain, diarrhoea). When these patients were grouped according to the different sites of mutations, mean values for age at onset of disease differed significantly: patients carryingAPC mutations at codon 1309 showed a disease onset 10 years earlier (mean age 20 years) compared with patients with mutations between codons 168 and 1580 (except codon 1309) (mean age 30 years), whereas patients with mutations at the 5′ end of codon 168 or the 3′ end of codon 1580 were diagnosed at a mean age of 52 years. Within each group of patients however large phenotypic variation was observed, even among patients with identical germline mutations. A higher incidence of desmoids was found in patients with mutations between codons 1445 and 1580 compared with mutations at other sites, while no correlation between site of mutation and presence of duodenal adenomas was observed.
CONCLUSIONS As age at manifestation and course of the disease may be rather variable, even in carriers of identical germline mutations, therapeutic decisions should be based on colonoscopic findings in individual patients rather than on the site of mutation. However, in patients with mutations within codons 1445–1580, it may be advisable to postpone elective colectomy because desmoids may arise through surgical intervention.
- familial adenomatous polyposis
- APC gene
- genotype-phenotype correlation
- phenotypic variation
Abbreviations used in this paper
- FAP
- familial adenomatous polyposis
- APC gene
- adenomatous polyposis coli gene
- AAPC
- attenuated adenomatous polyposis coli
- CRC
- colorectal cancer
- bp
- base pairs
Statistics from Altmetric.com
Familial adenomatous polyposis (FAP) is an autosomal dominant precancerous condition of the colorectum. In typical FAP, patients develop hundreds to thousands of adenomatous polyps. Colorectal symptoms such as rectal bleeding, diarrhoea, and abdominal pain occur in untreated patients at an average age of 29 years, and patients die from colorectal cancer (CRC) at an average age of 40 years.1 ,2 FAP is caused by germline mutations in the adenomatous polyposis coli (APC) gene which encodes a tumour suppressor protein consisting of 2843 amino acids.3 ,4 The APC protein is involved in downregulation of free intracellular β-catenin, a central component of the E-cadherin adhesion complex and the wntsignalling pathway.5
The majority of germline mutations identified in FAP patients are distributed over the 5′ half of the APCgene.6-11 Depending on the patients examined and the methods used for mutation analysis, germline mutations have been detected in 30–85% of FAP families. A correlation between site of mutation and clinical phenotype encompassing both colonic and extracolonic features has repeatedly been reported. Mutations at codon 1309 lead to the most severe intestinal phenotype characterised by a high number and early development of colorectal adenomas.2 ,12-14 A mild phenotype (attenuated FAP; AAPC) with smaller numbers of adenomas (<100) and a later age of onset was found to be associated with mutations localised either in the 5′ or 3′ part of the gene15-20 or in the alternatively spliced fragment of exon 9.11 ,21-24 Some of the extracolonic manifestations of FAP, including congenital hypertrophy of the retinal pigment epithelium,25-27 desmoids, and osteomas have also been correlated with the site of APCmutation.27 ,28
The discovery of the APC gene represents a breakthrough for carrier detection and surveillance in FAP families. While predictive testing among subjects at risk is now part of routine molecular genetic practice,9-11 ,29 the value of mutation analysis for therapeutic decisions is frequently overestimated among clinicians.
Based on 1256 patients with FAP who belonged to 680 families, we examined age of onset, course of colorectal polyposis, and clinically relevant extracolonic manifestations according to the germline mutation. The aim of the study was to see if the known genotype-phenotype correlation allows therapeutic conclusions to be drawn for the individual patient.
Methods
PATIENTS
Blood samples from 680 unrelated patients with a clear diagnosis of FAP or suspected of having FAP were examined forAPC germline mutations at the Institute of Human Genetics, Bonn. A total of 268 of the index patients presented with more than 100 colorectal adenomas; 101 patients had less than 100 adenomas. The number of adenomas was not known in 311 patients but most were reported to have “multiple adenomas characteristic of typical FAP” (table 1). Family histories revealed 357 index patients with at least one affected family member in previous generations. Fifty three patients seemed to have acquired a de novo mutation as both parents had negative colonoscopies at age >50 years or were still healthy at an advanced age. For 270 patients referred for genetic testing, no information on the parents was available or the parents had died at a young age from causes other than FAP (table 1). Clinical data of 1256 patients from the 680 families were obtained during genetic counselling or clinical management, or from medical records. We particularly documented whether the diagnosis of FAP was made after clinical bowel symptoms had developed (diarrhoea, rectal bleeding, abdominal pain) or through surveillance by endoscopy in subjects at risk. The study was approved by the local ethics committee.
APC germline mutations in 680 index patients grouped according to number of colorectal adenomas and family history
MUTATION ANALYSIS
Genomic DNA was isolated from peripheral blood using standard protocols.30 Total RNA was extracted from EDTA blood samples with Trizol (Life Technologies, Karlsruhe, Germany) according to the manufacturer's protocol. In the first 310 index patients, a search for mutations over the wholeAPC gene was performed with genomic DNA by non-radioactive single strand conformational polymorphism and heteroduplex analysis, as described previously,7 using the primers published by Groden and colleagues.3 In the remaining 370 index patients exon 15 was examined by protein truncation test in four fragments, essentially as described previously,31 with the in vitro transcription translation kit (Promega, Mannheim, Germany) in the presence of 35S methionine (Amersham). When mRNA was available, cDNA was obtained by standard procedures, and exons 1–14 were examined by the protein truncation test in two fragments using the primers previously described.32 Polymerase chain reaction fragments showing variant bands by either method were sequenced using Sequenase version 2.0 (Amersham) in the presence of 35S dATP or by semiautomated cycle sequencing with BigDye chain terminators with an ABI 377 sequencer (Applied Biosystems, Weiterstadt, Germany).
STATISTICAL COMPARISONS
Comparison of different clinical features in relation to the site of mutation were performed using the two tailedt test with unequal variance.
Results
MUTATION ANALYSIS
Pathogenic APC germline mutations were identified in 327 of the 680 index patients (48.1%). The mutation detection rate was 58.2% in patients diagnosed with >100 adenomas and 31.7% in patients with <100 adenomas (table 1). Remarkably, the mutation detection rate reached 59.7% in the 357 familial cases and 75.5% in the 53 patients with a de novo mutation.
Most of the mutations (322/327) were predicted to lead to truncated APC proteins, including 226 frameshift mutations due to small deletions/insertions, 87 nonsense mutations, and nine mutations in the highly conserved splice site sequences. In addition, four submicroscopic deletions uncovered through haplotype analysis in patients with normal behaviour and intelligence (Mandl and colleagues33 and unpublished observations), and one large cytogenetically detectable deletion of about 10 Mb encompassing the entire APC gene in a patient with mild mental retardation34 were observed. In accordance with previous reports, germline mutations were mainly distributed over the 5′ half of the APC gene (fig 1). A total of 170 different mutations were identified, including 86 novel mutations (table 2) not reported in the APC mutation data base (http://www.umd.necker.fr).

Distribution of APC germline mutations in 327 of 680 FAP families (47 of the 53 patients had the 5 bp deletion at codon 1309).
Novel APC germline mutations identified in 327 of 680 FAP families (mutations not reported in the APC mutation database)
Mutational hotspots published in other FAP patient populations6 ,9 ,10 were also observed in our patients. Deletion of 5 base pairs (bp) at codon 1309 was present in 47 of the 680 index patients (6.9%). All except one patient showed typical or very early onset FAP. The one patient who developed symptoms at age 53 had a de novo mutation. The unusually mild phenotype in this patient (100–200 adenomas without dysplasia at age 53) might eventually be due to a somatic mosaic. One of his sons who inherited the 5 bp deletion at codon 1309 was still asymptomatic at age 17; at colonoscopy performed following predictive genetic testing innumerable small polyps were detected. Another six patients had a 4 bp deletion or a nonsense mutation at codon 1309. Deletion of 5 bp at codon 1061 was detected in 33 index patients (4.9%), all of whom suffered from typical FAP. At least 17/47 mutations at codon 1309 and 5/33 mutations at codon 1061 had occurred de novo in the index patients, as demonstrated by exclusion of the mutations in both parents (data not shown).
PARAMETERS FOR SEVERITY OF COLORECTAL POLYPOSIS
Age at appearance of first colonic adenomas, and number of polyps and their progression during a defined interval of several years would be the most accurate parameter for studying the natural course of the disease. However, so far such data are available only for a small number of patients at risk who underwent regular colonoscopies starting at an early age. Age at clinical diagnosis of FAP in asymptomatic subjects at risk is largely dependent on age at first endoscopy and whether surveillance has been performed regularly. Therefore, we considered that age at diagnosis in this patient group was not suitable for comparison.
Further indicators of the severity of the natural course of bowel disease ascertained in a retrospective study are age at diagnosis of FAP in patients who have developed bowel symptoms characteristic of FAP (rectal bleeding, diarrhoea, etc) prior to their first endoscopy, and age at which CRC occurs in untreated patients. Therefore, we determined how the diagnosis of FAP or CRC was made.
Age at clinical diagnosis of FAP was known for 404 patients who had developed bowel symptoms prior to diagnosis and for 292 patients who were diagnosed on the basis of a surveillance programme offered to subjects at risk (table 3). As expected, mean age at diagnosis of FAP was lower in the surveillance group (25.4 years) compared with the group of patients who were diagnosed after they had developed symptoms (32.9 years).
Age at diagnosis in familial adenomatous polyposis (FAP) patients grouped according to the method of diagnosis
Overall, CRC was diagnosed in 193 of 1256 patients (15.4%), with a mean age of 40.7 years (table 3). A total of 126 of 410 patients diagnosed because of bowel symptoms (30.7%) developed CRC; 107 (26.2%) were found to have CRC at the time of diagnosis of FAP. In the group of 303 patients diagnosed while on surveillance, only 18 (5.9%) developed CRC; 11 (3.6%) presented with CRC at the time of diagnosis.
Similar data regarding age at diagnosis of FAP or CRC, respectively, were observed in the group of Danish FAP patients.1
As not all subjects at risk diagnosed during surveillance had undergone regular colonoscopies from a young age, clinical data from this group were not included in the genotype-phenotype analysis. Rather, age at diagnosis of FAP or CRC, respectively, was evaluated only in relation to the germline mutation in the group of 404 untreated patients diagnosed after spontaneous bowel symptoms had developed.
GENOTYPE-PHENOTYPE CORRELATION
According to the literature, attenuated FAP is associated with mutations 5′ to codon 168, 3′ to codon 1580, or in the alternatively spliced part of exon 9, and a very severe clinical phenotype is present in patients with mutations at codon 1309, while typical FAP is observed in patients with mutations at other sites. The germline mutation was uncovered in 230 of 404 patients who were diagnosed after onset of bowel symptoms. When grouping patients according to the mutation intervals outlined above, a consistent correlation between theAPC mutation site and average age at diagnosis of FAP was observed (table 4). Patients with a mutation at codon 1309 developed bowel symptoms more than 10 years earlier (19.8 years) compared with patients with other mutations: 52.2 years in patients with mutations at sites characteristic of attenuated FAP and 30.4 years in patients with mutations at other sites (p<0.0001 for all comparisons). This observation independently supports earlier findings.2
Relationship between mutation site and age at diagnosis achieved after bowel symptoms had developed in 230 patients with known mutations and in 174 patients with unknown mutations
Despite this clear genotype-phenotype relationship observed when mean values were compared, there was a wide variation in age at onset of intestinal symptoms within each of the subgroups, even among patients with identical mutations. Figure 2 presents age at diagnosis of FAP due to bowel symptoms in 230 individual patients from 205 families in relation to the site of their germline mutation. Only patients with mutations at codon 1309 (or close to this codon) were diagnosed before the age of 10 years. However, an appreciable number of patients with this mutation developed symptoms only at age 30–40 or even at age 53.
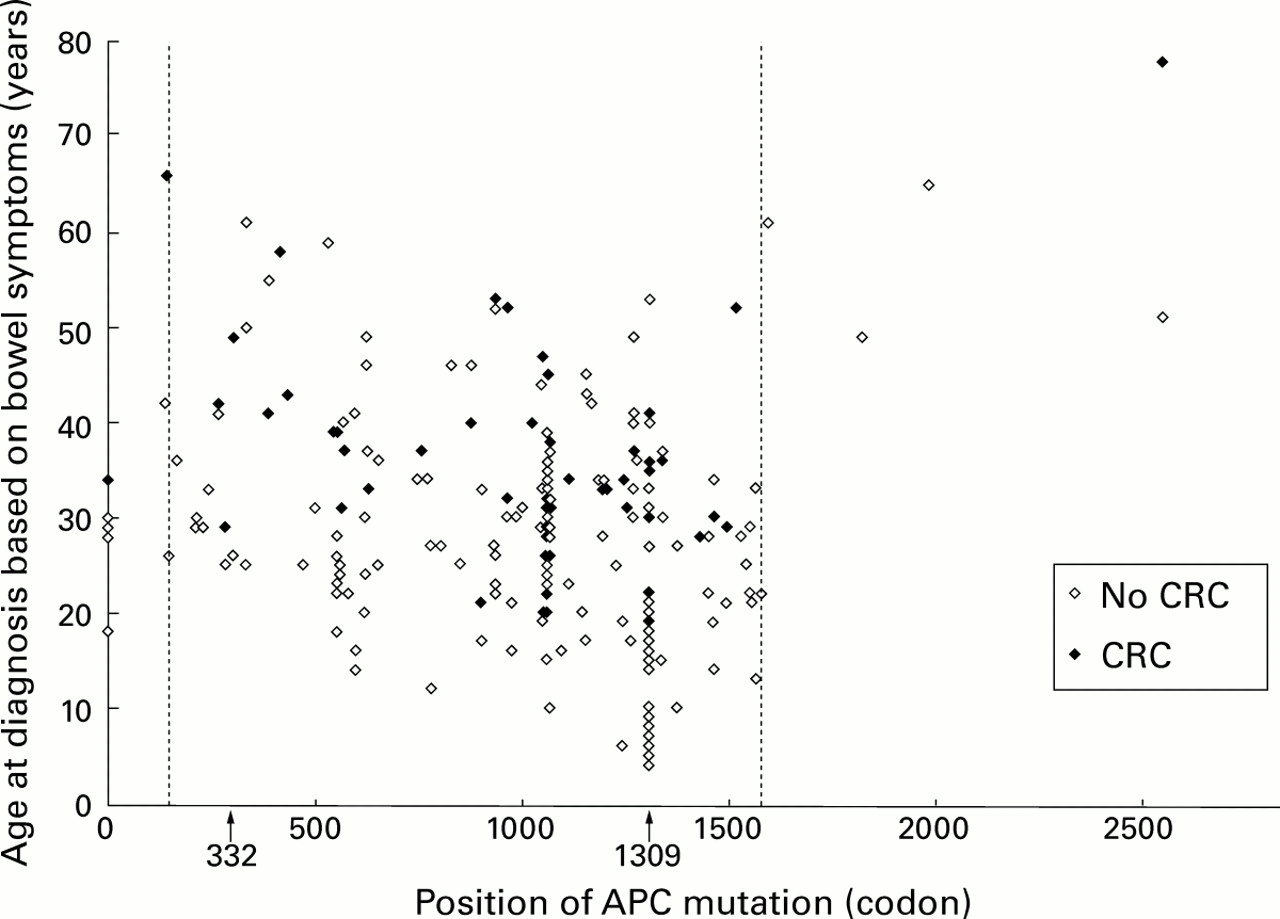
Age at diagnosis of familial adenomatous polyposis (FAP) due to bowel symptoms in 230 patients with identified germline mutations, in those in whom colorectal cancer (CRC) was present at diagnosis of FAP and in those without CRC. Mutations outside the vertical lines at codons 168 and 1580, and at codon 332 (in the alternatively spliced fragment of exon 9), respectively, are considered to be associated with attenuated FAP. Patients with large genomic deletions are depicted as “null alleles” at codon 0.
Large variations within patients with the same mutation were also observed when age at diagnosis of CRC in untreated patients was considered (figs 2, 3). The germline mutation was known in 97 of the 193 patients who developed CRC; in these 97 patients CRC was diagnosed at a mean age of 36.9 (0.9) years. CRC was diagnosed in 10 patients with the mutation at codon 1309 at a mean age of 28.8 years, with a large range between 19 and 41 years.

Age at diagnosis of colorectal cancer (CRC) in 97 patients with a known APC germline mutation. This group includes patients who had developed CRC at the time of diagnosis of familial adenomatous polyposis (FAP) (see fig 2) and patients who developed cancer one to several years after diagnosis of FAP.
CLINICALLY RELEVANT EXTRACOLONIC MANIFESTATIONS
Adenomas of the duodenum and desmoid tumours represent extracolonic features with important clinical implications. Adenomas of the duodenum were reported in 134 patients from 125 families. In 86 patients from 77 families the germline mutation was known, being localised between codons 156 and 1563, with a distribution that resembled the overall distribution of germline mutations in the index patients (fig 4). Thus no correlation between the presence of adenomas in the duodenum and site of mutations was evident.

Presence of duodenal adenomas in 86 patients with different germline mutations.
Information on the presence or absence of clinically manifest desmoid disease was obtained from 269 patients with identified mutations. Sixty five of these patients presented with desmoids (overall 24%). Although in patients with desmoids the APC mutations were spread over large parts of the gene, an uneven distribution was observed (fig 5). Desmoids were found in only 20% of patients with mutations 5′ to codon 1444 (46/230) but occurred in 49% (19/39) of patients with mutations 3′ to this codon and in 61% (17/28) of patients with mutations within codons 1445–1580 (fig 4, insert). Only two of 11 patients with mutations 3′ to codon 1580 developed desmoids.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Presence or absence of clinically manifest desmoid disease in 269 patients with identified germline mutations. The insert shows patients with mutations within codons 1445–1578.
Discussion
Examination of APC germline mutations in FAP families is being used as an efficient tool for predictive testing of subjects at risk. If the germline mutation causing the disease is known in a given family, it is possible to clearly distinguish between carriers who need to undergo surveillance by regular endoscopies and non-carriers who can be excluded from a surveillance programme.
Mutation analysis in large samples of FAP patients has revealed a consistent correlation between the site of mutation in theAPC gene and severity of intestinal polyposis or presence of clinically relevant extracolonic features.2 ,12 ,15 ,18 ,19 ,23 ,27 We too found this correlation in the present study. This relationship may be explained in part by recent studies demonstrating that the site of the germline mutation modulates the spectrum of somaticAPC mutations which lead to adenoma formation. Tumours from patients with attenuated FAP and a germline mutation at the 5′ end of the gene frequently maintain the wild-type allele35 whereas adenomas from patients with germline mutations close to codon 1300 display loss of the wild-type allele more frequently compared with patients with mutations at other sites, thus providing a greater selective advantage to these cells.36These findings may explain clinical observations in patients with the 5 bp deletion at codon 1309. Patients with this mutation tend to develop adenomas more frequently and at an earlier age than patients with other mutations.
An important question is whether the known genotype-phenotype relationship in FAP can be helpful for individual therapeutic decisions in either presymptomatic or symptomatic mutation carriers. Initial recommendations on application of molecular genetic tests as a guide to surgical management of FAP patients37 were based on the observation of a higher rate of rectal cancer in patients with mutations 3′ to codon 1250 compared with patients with mutations 5′ to this codon. The authors recommended a restorative proctocolectomy instead of a colectomy with ileorectal anastomosis for patients with mutations 3′ of codon 1250. However, this recommendation was later revoked38 ,39 as in the meantime several families with mild polyposis were found to have mutations in the 3′ half of the gene.17 ,40 ,41
Our results demonstrate a large variation with respect to both age at onset of intestinal symptoms and development of CRC even in patients with the same mutation (fig 2). In accordance with these data, variable phenotypes were reported within large FAP families.16 ,20 ,42 ,43 The observed variation may be caused by modifier genes or other endogenous or exogenous factors, or even by chance, as the “second hit”44 inactivating APC and inducing tumour growth occurs as a stochastic event. Thus the site of the APC mutation, although statistically correlated with the natural course of the disease, cannot predict its course in the individual patient.
Regarding clinically relevant extracolonic features, we found no correlation between the site of mutation and presence of duodenal adenomas. These results are in line with a recent study which reports a cumulative lifetime incidence for duodenal polyps of 97%.45 A higher incidence of desmoid tumours in patients with mutations located 3′ to codon 1444 compared with patients with mutations 5′ to this codon has consistently been reported by us and others,27 ,28 and was again confirmed in this study. However, not all patients with mutations 3′ to codon 1444 developed clinical desmoid disease although a subclinical manifestation cannot be excluded. Desmoids may develop particularly after surgical intervention. As at least 60% of patients with mutations within codons 1445–1580 develop desmoids, it seems advisable to postpone elective colectomy in patients with such mutations until disease progression in the colon is substantial. This decision requires the expertise of highly experienced clinicians.
In conclusion, identification of germline mutations in FAP patients is important for predictive testing in subjects at risk. Our analysis in a large number of FAP families showed that mutation analysis is of limited value for predicting the clinical course of the disease and for surgical decisions in individual patients, given the large phenotypic variation in patients with identical APCmutations. Therefore, decisions on clinical management have to be based on the degree of colonic polyposis. Nevertheless, other factors, including the presence of desmoids in the patient or in the patient's family, and their higher incidence in patients with mutations beyond codon 1444, also have to be taken into consideration. It is imperative that FAP families be referred for genetic counselling, genetic testing, early diagnosis, and clinical management to centres that are aware of the complexity of both surgical intervention and possible implications of molecular findings for individual patients.
Acknowledgments
The authors are grateful to the patients who participated in the study and to the many colleagues who contributed clinical data. The help of Vera Breisig for technical assistance is also kindly acknowledged. The study was supported by the Deutsche Krebshilfe within the national collaborative study on familial colon cancer.
Abbreviations used in this paper
- FAP
- familial adenomatous polyposis
- APC gene
- adenomatous polyposis coli gene
- AAPC
- attenuated adenomatous polyposis coli
- CRC
- colorectal cancer
- bp
- base pairs