Article Text

Abstract
Background and aims Colorectal cancer (CRC) is the second most frequent cancer in developed countries. Newfoundland has the highest incidence of CRC in Canada and the highest rate of familial CRC yet reported in the world. To determine the impact of mutations in known CRC susceptibility genes and the contribution of the known pathways to the development of hereditary CRC, an incident cohort of 750 patients with CRC (708 different families) from the Newfoundland population was studied.
Methods Microsatellite instability (MSI) testing was performed on tumours, together with immunohistochemistry analysis for mismatch repair (MMR) genes. Where indicated, DNA sequencing and multiplex ligation-dependent probe amplifications of MMR genes and APC was undertaken. DNA from all patients was screened for MUTYH mutations. The presence of the BRAF variant, p.V600E, and of MLH1 promoter methylation was also tested in tumours.
Results 4.6% of patients fulfilled the Amsterdam criteria (AC), and an additional 44.6% fulfilled the revised Bethesda criteria. MSI-high (MSI-H) was observed in 10.7% (n=78) of 732 tumours. In 3.6% (n=27) of patients, CRC was attributed to 12 different inherited mutations in six known CRC-related genes associated with chromosomal instability or MSI pathways. Seven patients (0.9%) carried a mutation in APC or biallelic mutations in MUTYH. Of 20 patients (2.7%) with mutations in MMR genes, 14 (70%) had one of two MSH2 founder mutations. 17 of 28 (61%) AC families did not have a genetic cause identified, of which 15 kindreds fulfilled the criteria for familial CRC type X (FCCTX).
Conclusions Founder mutations accounted for only 2.1% of cases and this was insufficient to explain the high rate of familial CRC. Many of the families classified as FCCTX may have highly penetrant mutations segregating in a Mendelian-like manner. These families will be important for identifying additional CRC susceptibility loci.
- DNA mismatch repair
- colorectal cancer
- familial adenomatous polyposis
- mutY homologue
- microsatellite instability
- cancer genetics
- cancer susceptibility
- cancer syndromes
- colorectal cancer genes
- colorectal carcinoma
Statistics from Altmetric.com
- DNA mismatch repair
- colorectal cancer
- familial adenomatous polyposis
- mutY homologue
- microsatellite instability
- cancer genetics
- cancer susceptibility
- cancer syndromes
- colorectal cancer genes
- colorectal carcinoma
Introduction
Colorectal cancer (CRC) is the second most frequent cancer in developed countries. Inherited susceptibility is responsible for ∼35% of all cases,1 but highly penetrant germline mutations account for <6% of all cases.2
Significance of this study
What is already known about this subject?
Mutations in at least 6 different genes can cause hereditary colorectal cancer (CRC) and these account for up to 6% of all CRC cases. There are 3 major pathways known to be responsible for CRC – CIMP, CIN and MSI. However, there are likely additional pathways leading to hereditary and sporadic CRC.
What are the new findings?
We have shown that 3.6% of our population-based CRC study cohort has highly penetrant germline mutations in 6 different genes. There is a surprisingly high proportion of AC1 families that do not have a known genetic predisposition and thus were categorized as FCCTX. Patients from FCCTX families had a later age of CRC diagnosis and their tumours were more often located in the distal colon than in patients with Lynch syndrome.
How might it impact on clinical practice in the foreseeable future?
In the very near term, a population-based panel of specific mutations will be used to enhance CRC screening programs. Also, the FCCTX families we have identified in this study may segregate mutations in highly penetrant novel genes. Once identified, these genes should aid in the diagnosis of CRC as well as provide insights into more personalized treatments of CRC, in the same way that identifying the mismatch repair genes and their role in MSI has helped clinical practice.\
Although there have been many studies reporting inherited mutations in patients with CRC, to our knowledge there have been no published studies of such a comprehensive nature. In the current study, we have investigated an incident cohort of patients with CRC from a population of northern European descent and have tested for mutations in all six of the known major susceptibility genes. The research questions asked in the current report include the following. (1) What is the contribution of mutations in known CRC-related genes to the high incidence of CRC in the population? (2) Do founder mutations contribute to the high incidence of familial CRC in the population? (3) What is the phenotype of CRC arising from families at high risk of CRC of unknown genetic aetiology?
Materials and methods
The Newfoundland Colorectal Cancer Registry (NFCCR)
The NFCCR is modelled after the NIH-supported Colorectal Cancer Family Registries and specifically the Ontario Familial Colorectal Cancer Registry.10 11 Patients were eligible for the study if diagnosed with colorectal carcinomas (pure adenomas not included) between 1 January 1999 and 31 December 2003 and were under 75 years of age when diagnosed. Consenting patients received a family history questionnaire and were asked to provide a blood sample and permit access to tumour tissue and medical records. If a patient was deceased, we sought the participation of a close relative for the purposes of obtaining the family history and for permission to access tissue blocks and medical records. Use of proxies in this way removes the bias of excluding advanced-stage cancer patients who die before they can give consent.
Risk assessment was carried out as described by Green et al.12 Patients meeting the AC1 or Amsterdam criteria 2 (AC2)13 14 or our FAP criteria (the proband was previously known to be in FAP or attenuated FAP (AFAP) families with a mutation detected in APC, or clinical diagnosis of FAP or AFAP in the proband's family, or proband has >100 adenomatous polyps) were designated as high risk. Those meeting at least one of the revised Bethesda criteria (criteria #1, 2, 4 and 5 were considered only),15 and not meeting the high risk criteria, were designated as intermediate risk, and the remaining patients were considered as being at low risk for an inherited CRC syndrome. All participants in the study were provided with information on their risk classification, by letter, phone call or ‘in person’ counselling. Clinical screening recommendations were provided appropriate to their risk level.
Ethics approval was obtained from the Research Ethics Board of the Memorial University of Newfoundland, and all patients or their proxies provided informed consent.
Molecular testing procedure
To determine the contribution of the common pathways that might influence the development of CRC in our cohort, we performed a battery of assays on various biological sample types (figure 1A). In order to determine the involvement of the MMR genes and to help target the appropriate genes for downstream DNA sequencing, we extracted tumour DNA and performed MSI testing and immunohistochemistry (IHC) analyses for MLH1, MSH2 and MSH6. For tumours that were MSI-high (MSI-H), thus indicating an MMR deficiency, but not deficient in MLH1, MSH2 or MSH6, we then performed IHC for PMS2. Also, tumours were tested for MLH1 promoter methylation and the presence of the BRAF variant c.1799T→A (p.Val600Glu).

(A) The biological samples obtained from study participants, their derivatives and assays implemented. (*in a few cases RNA was extracted to confirm multiplex ligation-dependent probe amplification (MLPA) results). (B) Testing flow chart to determine mismatch repair (MMR) deficiency and Lynch syndrome. Numbers represent tumours/patients. The number of tumours at the top of the chart are those which had microsatellite instability (MSI) analysis performed. 1N.D., not determined; 2MLH1-D, MLH1-deficient and includes both MLH1-only and MLH1- and PMS2-deficient tumours; 3Meth or BRAF, methylated MLH1 promoter or presence of p.V600E BRAF mutation.
In cases with MSH2, MSH6 or PMS2-only deficient tumours and cases whose tumours were MLH1 deficient but lacked MLH1 promoter methylation, all of which were MSI-H, leucocyte DNA was tested for large duplications/amplifications by multiplex ligation-dependent probe amplification (MLPA). For individuals with tumours deficient in MMR proteins, the corresponding gene(s) (MLH1, MSH2, MSH6 and PMS2) were sequenced, except that MLH1 was only sequenced if the patient's tumour did not exhibit MLH1 promoter methylation. In order to ensure that high risk individuals definitively did not carry germline mutations in the commonly mutated MMR genes, all probands from AC1 and AC2 families were sequenced for MLH1, MSH2 and MSH6, and MLPA on MSH2 and MLH1 was performed regardless of the results of the preceding tests.
DNA from patients fulfilling our FAP criteria was tested for the APC mutations previously observed in Newfoundland. If one of these mutations was not identified in the patient, a more rigorous screening of APC was conducted (see below). To determine the frequency of autosomal recessively (eg, biallelic mutations) inherited MAP in our cohort, DNA from all patients was tested for MUTYH mutations.
Colorectal tumour analyses
IHC and MSI analyses
Our protocol for MSI analysis and IHC staining of the MLH1, MSH2, MSH6 and PMS2 proteins in colorectal tumours has previously been described.16 MSI status was assigned as MSI-H (≥30% of markers tested unstable), MSI-low (MSI-L, >0% and <30% of markers unstable) or microsatellite stable (MSS, no unstable markers).
MLH1 promoter methylation and BRAF (c.1799T→A) mutation analyses
Methylation was detected using the MS-MLPA kit ME001B (MRC-Holland, Amsterdam, The Netherlands). This kit can generate two methylation-dependent signals from the MLH1 promoter. We scored the tumour DNA sample as methylated if either of these fragments was present at a normalised ratio of 0.15 of the peak area of the sample before digestion with HhaII.17
To identify the c.1799T→A (p.Val600Glu) variant in BRAF, we used a protocol previously described.18 This is an allele-specific PCR assay which includes a set of primers for GAPDH (glyceraldehyde phosphate dehydrogenase) as an internal positive control.
Germline mutation screening
All nomenclature used to describe sequence variants conforms to the recommendations found on the Human Genome Variation Society website (http://www.hgvs.org), updated as of October 2007.
DNA sequencing of MLH1, MSH2, MSH6 and PMS2
Alterations of MLH1, MSH2 and MSH6 were determined by sequencing all 45 exons and intron/exon boundaries as described previously.16 The PMS2 (RefSeq NM_ 000535.4) variants were detected as described previously,19 with the modifications explained in Clendenning et al20.
Rearrangements within MLH1 and MSH2
Exon deletions and duplications in MSH2 and MLH1 were detected by MLPA21 in germline DNA as previously described.16 All rearrangements identified by MLPA were confirmed in other affected relatives by MLPA and, when possible, cDNA analyses.
DNA screening of MUTYH and APC
MUTYH testing was performed on DNA from 539 of the 552 patients (97.6%) who provided a blood sample. Denaturing high-performance liquid chromatography (dHPLC) (Transgenomic Wave 3500HT System, Omaha, Nebraska, USA) was used to detect MUTYH mutations, and all mobility shifts were confirmed by DNA sequencing as described previously.22
Screening APC was outsourced to either the Mayo Clinic (Rochester, Minnesota, USA) or the University of Calgary (Alberta, Canada). Two patients were from a family known to carry an APC splice site mutation (c.221-1G→A) causing FAP.23 These were tested in Calgary, Alberta by direct DNA sequencing of the relevant exon. A third patient was tested at the Mayo Clinic by protein truncation testing for exon 15, direct DNA sequencing of exons 1–14 and 5′ of exon 15, and by MLPA of the entire gene.
Statistical analyses
Comparisons between the age at diagnosis and risk category were performed using analysis of variance (ANOVA). Post hoc multiple comparisons were determined using the Tamhane–Hochberg procedure.24 Differences between age categories of patients and participation rates were performed by means of Pearson χ2, with Bonferroni correction for multiple comparisons. The Tamhane–Hochberg procedure was used when equal variances could not be assumed, and the Bonferroni correction was used when equal variances could be assumed. These assumptions are determined based on the significance of the Levene statistic which is a test for the homogeneity of variance. ANOVA was also used to compare age of diagnosis and mutation status. Bonferroni was used for post hoc testing. The t test was used to compare the age of diagnosis between patients with FCCTX and MMR mutations. Tumour site and mutation status were compared using Pearson χ2. Two-sided 95% CIs of the proportions of mutation carriers were estimated using Wilson score method without continuity correction.25 Differences in proportions were compared using the χ2 test. All tests were performed using the Statistical Package for Social Sciences (SPSS, Chicago, Illinois, USA) version 15.0.
Results
Family history
Of 1173 eligible patients or proxies, 750 (64%) from 708 different families consented to be in the study. The mean age of CRC diagnosis of all 750 cases was 60.6 years (figure 2) and 61% (457) of the patients were male. There was a significant difference between the ages at diagnosis of patients from families fulfilling Amsterdam criteria or revised Bethesda criteria compared with those patients from low risk families (p<0.001; p<0.001). Thirty-one families had two patients, four families had three patients and one family had four patients diagnosed with CRC over the 5 year period of the study. Family history information was available for 728 patients from 685 families. As shown in figure 3, 5.6% of the patients with family history data met our high risk criteria, 44.6% met our intermediate risk criteria and 49.8% were low risk.

Summary data from the Newfoundland Colorectal Cancer Registry (NFCCR). (A) Average age of diagnosis of colorectal cancer (CRC) for each risk category. Amsterdam criteria families are excluded in the revised Bethesda criteria category. (B) Participation rates of patients with CRC per age categories. Proxies are included and are assigned age at diagnosis of CRC for their affected relative. (C) Average age at diagnosis of CRC for mutation carriers and for familial CRC type X (FCCTX). (D) Proportion of tumours located in proximal and distal colon in mismatch repair (MMR)-deficient tumours and in tumours from patients in families fulfilling FCCTX. Proximal is defined as proximal to the splenic flexure. MMR refers to tumours from patients which have mutations in MLH1, MSH2, MSH6 or PMS2. Non-MMR refers to tumours from patients with biallelic MUTYH mutations or an APC mutation.

Genes mutated in patients from the Newfoundland Colorectal Cancer Registry (NFCCR) according to clinical risk criteria (*revised Bethesda criteria and excluding high risk families).
MSI and IHC
There were 772 colorectal tumours available from the 750 consenting cases (figure 4). MSI analysis was completed on 732 (94.8%) of the tumours: 78 (10.7%) were MSI-H, 31 (4.2%) were MSI-L and 624 (85.3%) were MSS (figure 1B; figure 4). The 78 MSI-H tumours were from 75 different patients: three patients had two synchronous colorectal tumours discordant for MSI status—one MSS and one MSI-H. Of the MSI-H tumours, 72 (92.3%) were deficient in at least one MMR protein. Our MSI results correlate well with the mutation results in that all patients with mutations in APC or MUTYH (biallelic) had MSS tumours, while all of the tested MMR mutation carriers had MSI-H tumours. The mean age of CRC diagnosis in patients with MSS tumours was 61.0 years compared with 58.2 years for those who had MSI-H tumours (p=0.02). The location of the tumour was also statistically significant (p<0.001), with MSI-H tumours more often proximal compared with the more commonly distal MSS tumours. The 30 tested MSI-L tumours were intact for all tested proteins using IHC and they appeared to be very similar to MSS tumours when comparing tumour location (p<0.001) with MSI-H tumours. However, there was no significant difference in age at CRC diagnosis (p=0.157) between individuals with MSI-L and MSI-H tumours. There was also no significant difference in the age at diagnosis between patients with MSS tumours and those with MSI-L tumours (p=0.786). None of the patients with MSI-L tumours was from a high risk family.

Summary of microsatellite instability (MSI) and immunohistochemistry (IHC) results from the 772 tumours collected through the Newfoundland Colorectal Cancer Registry (NFCCR). ‘IHC-I’, IHC-intact; ‘IHC-D’, IHC-deficient; ‘IHC-ND’, IHC not done.
Promoter methylation of MLH1 and BRAF status in colorectal tumours
The MLH1 methylation status of 75 of the 78 MSI-H tumours was determined, and 42 (56.0%) were methylated. We were able to determine both MLH1 methylation and the BRAF mutation status in 68 MSI-H tumours. Of these, in the 40 MLH1-methylated tumours tested, 31 (77.5%) had a BRAF mutation, whereas in the 28 MLH1-unmethylated tumours tested, just two (6.9%) had a BRAF mutation (p<0.001). None of the mutation carriers tested had a tumour with the BRAF mutation or with MLH1 methylation. Although there were only nine MLH1-methylated tumours that were not BRAF mutation positive, it indicates that BRAF is not a precise surrogate for MLH1 methylation in MSI-H tumours.
Mutations causing hereditary CRC susceptibility
From the 750 patients enrolled in this study, we identified 27 patients (3.6%) whose CRC could be attributed to mutations in known CRC-related genes (figure 3; table 1). Twenty patients (2.7%) had mutations in MMR genes; 3 (0.4%) had mutations in APC; and 4 (0.5%) had biallelic mutations in MUTYH.
Summary of tumour and DNA analyses of cases with causative mutations in known hereditary colorectal cancer genes
Of the 20 cases with MMR mutations, 14 (70%) had one of two founder mutations in MSH2. Two different mutations were also identified in each of MLH1, MSH6 and PMS2, with a single case associated with each mutation. Of the eight different MMR mutations identified in this cohort, six had been previously included in a variant database for MMR genes in Lynch syndrome families26 and two were novel mutations (MLH1, c.208-?_380+?dup; and MSH6, c.3557-1G→A).
Two patients with mutations in APC were from families previously known to have a founder mutation.23 The third APC mutation (in patient SN1049) was a previously documented nonsense mutation in exon 16.27 Two of the mutations identified in MUTYH are the most commonly reported in Caucasian populations: c.494A→G (p.Tyr165Cys) and c.1145G→A (p.Gly382Asp).22 28 Four patients clinically identified as having FAP were carrying homozygous MUTYH mutations (0.74% of 539 tested) and 14 patients (2.6%) were heterozygous for MUTYH mutations (Supplementary table 1). Of note, one patient (SN2290) carrying a heterozygous MUTYH (p.Gly382Asp) mutation also had a PMS2 mutation. No MUTYH mutations were identified in the MSH6 mutation carriers, supporting the hypothesis that there is no association between these genes.29
In summary, of the 78 tumours that were MSI-H, 42 (53.8%) showed methylation of the MLH1 promoter and a further 21 tumours (26.9%) were from patients carrying a germline mutation in an MMR gene. In the remaining 15 tumours (19.2%), no explanation for the deficiency in MMR was identified. Whenever genomic DNA was available (11 of 15), the corresponding gene(s) indicated to be deficient in IHC testing was sequenced and MLPA for MSH2 and MLH1 was performed.
Fifty-four non-pathogenic variants were identified in MLH1, MSH2 and MSH6 (Supplementary table 1). These are not considered the primary genetic cause of the disease in these cases, since most of these variants have been identified previously and evidence from the literature suggests they are not highly penetrant variants.
Genetic basis of CRC in AC1 families
Of the 32 patients from 26 families fulfilling AC1, 15 (46.9%) patients from 11 families had an MMR mutation identified (table 1). The remaining 17 patients in 15 AC1 families did not have a genetic cause identified, and these families were considered FCCTX. In all but one of the FCCTX families, patients had CRC that was MSS, BRAF mutation negative and no MLH1 methylation was identified. The exception had an MSS tumour without methylation but had the BRAF mutation. The age at diagnosis of patients categorised as FCCTX was significantly higher than those of patients with MMR mutations (figure 2C; p<0.011) and their tumours were more often located in the distal colon when compared with patients with MSI-H tumours (figure 2D; p<0.001).
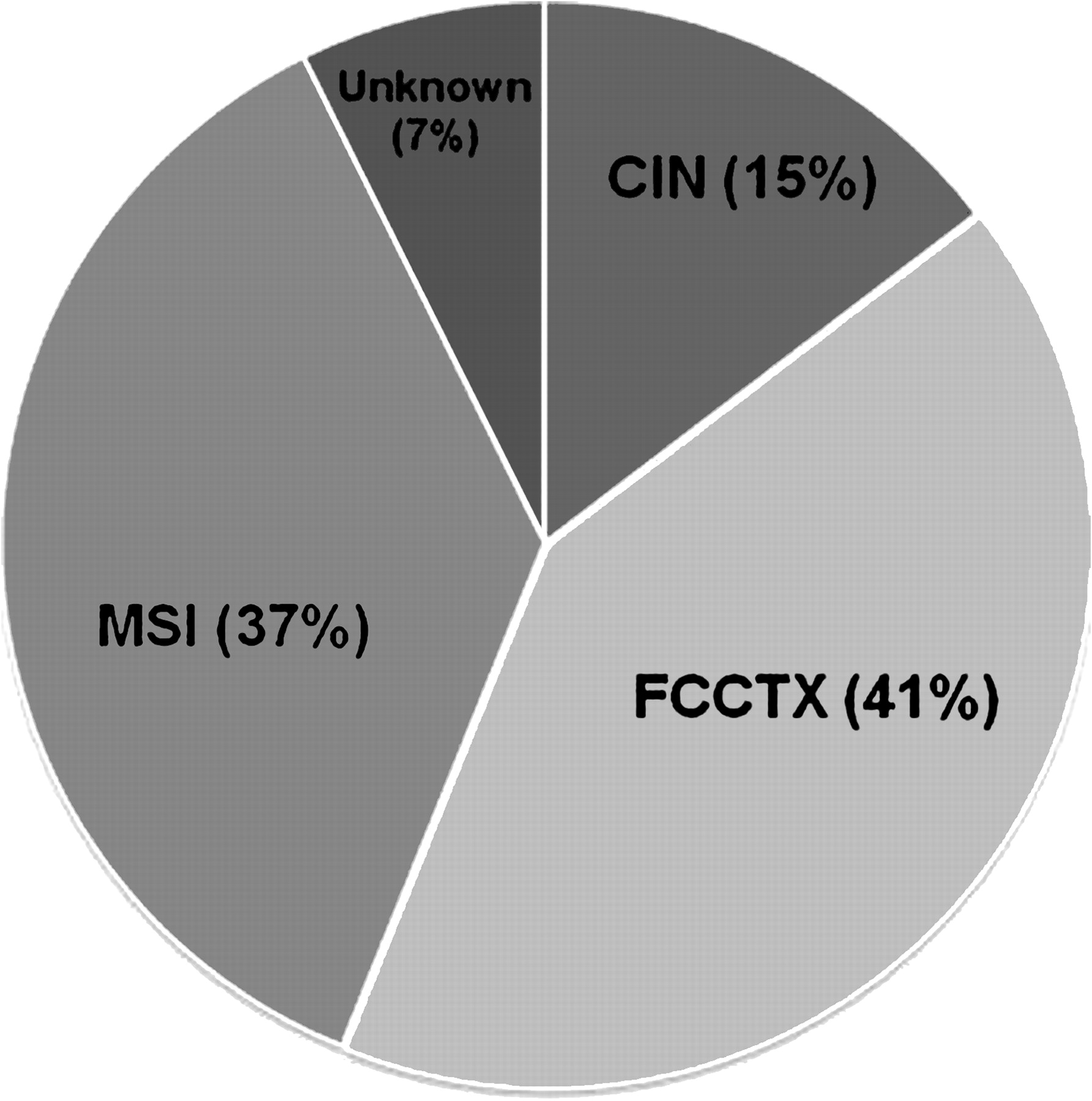
Figure 5 illustrates the contribution of major pathways predisposing to hereditary CRC in patients from high risk families. Inherited mutations in genes associated with CIN or MSI accounted for 52% of hereditary CRC. No AC1 patient had a CRC with both CIMP abnormalities (BRAF mutation and methylation of MLH1) while a large proportion (41%) were included in the FCCTX category.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proportion of patients (n=41) from high risk families indicating the genetic aetiology of their colorectal cancer predisposition. CIN, chromosomal instability; FCCTX, familial colorectal cancer type X; MSI, microsatellite instability.
Discussion
The population of Newfoundland (510 000) is a relatively new founder population almost exclusively of English and Irish descent.30 Importantly, of 12 founder populations described, Newfoundland has the best generalisability to other Caucasian populations.31 Newfoundland has the highest incidence of CRC in Canada and one of the highest rates of familial CRC in the world.12 The high rate of familial CRC in Newfoundland is demonstrated by the fact that >50% of incident cases fulfil Amsterdam or revised Bethesda criteria. This high rate, however, does not appear to be due to preferential recruitment of patients with a family history. We found that there was no significant difference (p=0.571) when we compared the proportion of cases which were eligible for participation who were <50 years of age (11.5%) with the proportion of recruited cases <50 years of age in our series (12.5%). Therefore, a bias in favour of young age, a predictor of hereditary CRC, did not occur in our series. Also, we are not aware of any other potential biases. Our recruitment rate of 64% is a true ascertainment rate for the population under study. Considering this is a retrospective study, all eligible cases included the deceased, and our initial contact with the patients was through their family physicians (due to Research Ethics Board requirements), this rate is quite high.32 33 Our study's recruitment rate may have increased if we recruited at the time of diagnosis or at time of surgery.
There were 12 different mutations accounting for 3.6% (95% CI 2.5% to 5.4%) of the patients with CRC from this population. Three founder mutations were observed in this population, one in APC and two in MSH2.28 34 The most common Lynch syndrome-associated mutation in the world (MSH2 c.942+3A→T) was identified in 10 patients. In Newfoundland, all carriers of this mutation have a common haplotype and a common geographic origin, indicating a founder effect, whereas the increased frequency worldwide is due to recurrence of the mutation.35 The total proportion of MMR mutations was 2.7% (95% CI 1.7% to 4.2%), which too is noteworthy, as it is very similar (2.8%) to that found in a thorough multihospital study carried out in the USA.36 This is important as it indicates that the Newfoundland population is comparable with North American and European outbred Caucasian populations in terms of the burden of known hereditary CRC predispositions in the population. Likewise, we identified 0.74% (95% CI 0.29% to 1.89%) of our cases as having biallelic mutations in MUTYH, not dissimilar to the findings of a large population-based study from Scotland which found that 0.8% of cases aged <55 years and 0.54% of the entire cohort had biallelic mutations.37
Of the 27 mutation carriers, 19 had tumours which were MSI-H and 21 belong to families with a high risk of CRC. In tumours from 42 additional patients, we observed methylation in the MLH1 promoter, loss of MLH1 staining and/or MSI, suggesting that this phenomenon was an important mechanism in the aetiology of disease in these patients. Thus 61 of 78 (78.2%) patients with MSI-H tumours could be linked to a specific gene (epi)mutation. None of the tested MSI-H tumours from carriers of MMR mutations showed MLH1 methylation, and methylation was not the second hit in either of the two MLH1 mutation carriers.
However, there were 15 of 78 (19.2%) tumours that were MSI-H but were not MLH1 methylated, yet no mutations in MMR genes could be identified in the 11 patients in which germline DNA was available. Perhaps mutations in the corresponding genes were not identified in this group of patients with suspected Lynch syndrome due to the screening protocol, as mutations could have occurred deep within introns or in upstream regulatory regions, both of which were not entirely screened. The TACSTD1 gene and the region between MSH2 and TACSTD1, which has recently been implicated in Lynch syndrome, was not screened in patients with MSH2-deficient tumours, and we were not able to determine the occurrence of MLH1 germline epimutations.38 39
In addition, the majority of AC1 families did not have a mutation identified in the six genes extensively examined. These families were therefore categorised as FCCTX and none had evidence of methylator pathway dysfunction. Tumours from the probands of the high risk families that did not have a mutation identified were MSS and had MMR proteins intact. This suggests that, in our cohort, there may be additional highly penetrant mutations of novel CRC-predisposing genes. There is strong evidence in the literature that other CRC-predisposing genes exist.40–44 CRC loci recently identified using genome-wide association studies most probably involve low penetrance mutations44 and it is possible that these loci, in combination with other low penetrance loci,40 may contribute to the cause of cancer in our cohort of FCCTX families.
As has been observed previously,45 46 MSI-L tumours do not appear to be associated with defects in the MMR pathway: none of our MSI-L tumours had deficiencies in MMR protein expression. Approximately half of our MSI-L tumours (16/31) exhibited instability of a dinucleotide marker, with the remaining 15 displaying instability of a mononucleotide marker. However, all of the 78 MSI-H tumours showed instablility for at least one mononucleotide marker and the vast majority (74/78) were unstable for two mononucleotides (for four there were incomplete data). Indeed, MSI-L status may not be associated with any clinical or molecular variable but may be a result of normal somatic slippage at microsatellite loci.47 Therefore, we suggest, as others have,48–50 that mononucleotide markers alone could be used to determine MSI.
In a number of cases our IHC data were not consistent with the MSI or mutation status, which is not uncommon.51 In an MSH6 mutation carrier (SN1002, table 1), the tumour appeared to be deficient in both MSH2 and MSH6. In one patient (SN1303) with an MSH2 mutation, the IHC testing indicated a deficiency in MSH6, with apparently normal MSH2 protein staining. In another MSH2 mutation carrier (SN2150), IHC testing identified a deficiency in both MLH1 and MSH6 proteins, but not MSH2. Anomalies were also observed in patients lacking MMR mutations. For example, two patients without MMR mutations had both MLH1- and MSH6-deficient tumours and in one of these patients (SN2001) the tumour was MSI-H and the MLH1 promoter was methylated, yet no mutation was identified in MSH6 following sequencing. In the second case (SN1826), the tumour was MSS, there was no MLH1 methylation detected and no mutation was found. These tumours were tested more than once with the same results. Another unusual IHC finding was an MSI-H tumour which had deficiencies in all four proteins tested (SN2425). Unfortunately, DNA was not available from this patient so mutation testing could not be performed. At least some of these ambiguities may be associated with poor fixation of the tumours.52
The Provincial Medical Genetics Program, which is responsible for all genetic screening in the region, has an algorithm for detection of mutations in CRC families, with the initial separation of ‘polyposis’ and ‘non-polyposis’ CRC families to be considered for APC/MUTYH and MMR mutations, respectively. However, from the mutation analysis in the population-based study, there is not always a clear division between these two groups. Through this study we have identified a number of patients with an APC mutation or MUTYH biallelic mutations that differ substantially in the number of polyps observed at diagnosis (see table 1). As reviewed by Poulsen and Bisgaard in 2008,53 our results are similar to previous studies in that patients with biallelic MUTYH mutations usually do not present with classical FAP (>1000 polyps) but are rather more similar to patients with AFAP (3–100 polyps) at diagnosis. Also, we did observe one patient (SN1311) with MUTYH biallelic mutations who had no polyps upon CRC diagnosis. Such an observation is not unique; however, it is uncommon.22 Our two APC mutation carriers were at the extreme ends of the FAP spectrum, with one having >1000 polyps and the other <20 polyps. Thus, this latter case overlaps phenotypically with some of our patients with MAP and even some of our patients with Lynch syndrome in regards to polyp count. Although not performed in our study, screening tumours for the c.34G→T variant in KRAS2 is a cost-effective method of identifying atypical MUTYH mutation carriers such as patient SN1311. This somatic mutation has been shown to be more common in patients with MAP compared with those with sporadic CRC.51
We have assembled an incident cohort of 750 patients diagnosed with CRC, from the population of Newfoundland, over a 5 year period. The only exclusion criterion was age ≥75 years. We have performed a comprehensive analysis to identify inherited mutations in two genes associated with CIN (APC and MUTYH) and the four major genes associated with MSI (MSH2, MLH1, MSH6 and PMS2). In addition, we identified the contribution of FCCTX and cancers associated with methylator pathway abnormalities in high risk families. To date, this is the most thorough population-based study of the genetic aetiology of CRC. Furthermore, founder mutations accounted for only 2.1% of cases and this was insufficient to explain the high rate of familial CRC. Due to the population structure and the similarity of the environment shared by most Newfoundland families, it is possible that the genetic aetiology of CRC in some of our FCCTX families is due to highly penetrant mutations segregating in a Mendelian fashion. Additional studies are needed to determine the basis of CRC susceptibility in these families.
Acknowledgments
We would like to express our deepest thanks to the families and patients who have so generously provided their time and biological samples to the NFCCR. We also thank Leigha Senter, MS, CGC, Ohio State University for her effort regarding the PMS2 mutation screening, and all of the staff associated with the NFCCR who rigorously prepared some of the data for analyses (Angela Batstone, Debbie Carroll, Sarah Mathieson, Carol Negrijn and Barbara Smith).
References
Supplementary materials
Web only data gut.2010.208462
Files in this Data Supplement:
Footnotes
Funding Supported by the Canadian Institutes of Health Research grant CRT-43821; National Cancer Institute of Canada grants 18223 and 18226; Genome Canada (Atlantic Medical Genetics and Genomics Initiative); National Cancer Institute (USA) grant CA16058.
Competing interests None.
Ethics approval This study was conducted with the approval of the Memorial University of Newfoundland, St. John's, NL, Canada.
Provenance and peer review Not commissioned; externally peer reviewed.