Article Text

Abstract
Mutations in the gene encoding Cu/Zn superoxide dismutase (SOD1) account for approximately 20% of patients with familial amyotrophic lateral sclerosis (FALS). In this study, sequence analysis of exons 1–5 of SOD1 in a large German cohort with FALS was performed. Among 75 affected patients, who were not obviously related probands with a positive family history, nine had missense mutations in SOD1. Four of the nine probands carry the same R115G mutation in exon 4 of the SOD1 gene. Genotyping with markers from the SOD1 locus revealed a common haplotype and shared allelic characteristics in these patients. These findings suggest that the R115G mutation in the German population originates from a common founder.
- ALS, amyotrophic lateral sclerosis
- FALS, familial amyotrophic lateral sclerosis
- PCR, polymerase chain reaction
- SNP, single nucleotide polymorphism
- SOD1, Cu/Zn superoxide dismutase
- amyotrophic lateral sclerosis
- SOD1
- founder mutation/effect
- haplotype
- mutational analysis
Statistics from Altmetric.com
- ALS, amyotrophic lateral sclerosis
- FALS, familial amyotrophic lateral sclerosis
- PCR, polymerase chain reaction
- SNP, single nucleotide polymorphism
- SOD1, Cu/Zn superoxide dismutase
Amyotrophic lateral sclerosis (ALS) is a fatal, degenerative disorder of the brain and spinal cord motor neurones, which results in progressive paralysis. Although most cases are sporadic, 5–10% of patients have a positive family history.1 Familial ALS (FALS) is genetically heterogeneous and eight loci and two disease genes have been identified.2,3 FALS is mostly inherited as an autosomal-dominant trait. Autosomal-dominant and sporadic forms are frequently clinically similar. Disease causing mutations in the gene encoding Cu/Zn superoxide dismutase (SOD1) on chromosome 21q22.1 account for approximately 15–20% of cases of FALS.4,5 More than 100 different SOD1 mutations in all five exons and at splice sites of SOD1 have been identified in FALS.
In our study, we performed an extensive mutational analysis in 75 probands with a positive family history of ALS by sequencing all five exons of SOD1. We provide evidence for a common founder of the R115G mutation in the German population. Furthermore, we determined the proportion of patients with FALS as a result of mutations in SOD1.
METHODS
Patients
In total, 75 patients from Germany (n = 71), Austria (n = 2), and Switzerland (n = 2) were recruited for our study. All participants were white, of European ancestry, and not known to be related. The patients were ascertained at academic neurological departments and the diagnosis of ALS was established by clinical and electrophysiological criteria. All patients met the following criteria: (a) a diagnosis of probable or definite ALS6 and (2) a positive family history with at least one affected first or second degree relative. To exclude patients with possible recessive modes of inheritance, we did not include affected sibpairs without a parental history of ALS or other affected first or second degree relatives. Information about age at onset and family history was obtained by direct interview.
Molecular studies
Peripheral blood was collected after informed consent was given, and genomic DNA was extracted using the QIAamp DNA blood kit (Qiagen, Hilden, Germany). SOD1 exons 1–5 (complete cDNA sequence) including flanking intronic sequences were amplified by polymerase chain reaction (PCR). This was followed by direct sequencing of both strands of PCR products on an ABI-Prism 3100 DNA sequencer, and subsequent sequence analysis using the SeqAnalysis (version 3.7) and SeqScape (version 2.0) software.
Intragenic single nucleotide polymorphisms (SNPs) were ascertained by sequence analysis for both the established SNP in intron 3 (IVS3+34A>C)7 and a newly identified SNP in intron 1 of SOD1 (IVS1-108T>A).
The short tandem repeat polymorphisms (STRPs) D21S263, D21S63, D21S1252, and the new microsatellite “Yorick”8 were genotyped by [α32-P] dCTP labelled PCR using the published primer sequences (Genome Database (http://www.gdb.org)) and analysed by polyacrylamide electrophoresis followed by autoradiography. Allele sizes were determined by comparison with an M13 sequence ladder.
The order of SNPs and STRPs (centromere to telomere) was: D21S263, Yorick, SNP SOD1 intron 1 (IVS1-108T>A), SNP SOD1 intron 3 (IVS3+34A>C), D21S63, and D21S1252.
Control allele frequencies were determined by genotyping 90 unrelated individuals from the same ethnic background. Haplotypes were constructed manually allowing for a minimum number of recombinations.
A statistical analysis of allelic association between the R115G mutation and marker alleles from the common haplotype was performed based on the one tailed Fisher’s exact test for a 2 × 2 table. For the STRPs and SNPs, association was tested by comparing the frequency of alleles in the four R115G probands and alleles in the 90 controls. A p value of ⩽ 0.01 was considered significant for association.
RESULTS
We identified six different SOD1 mutations in nine of the 75 not obviously related probands analysed (table 1). All mutations were missense mutations in the open reading frame of SOD1 exons 2, 4, and 5 and have been reported before (see ALS mutation database (http://www.alsod.org)). In addition, we detected a T → A substitution in SOD1 intron 1, 108 base pairs upstream of exon 2 (IVS1-108T>A) in both patients and controls. Mean age at onset in patients with a SOD1 mutation was 52 years (range, 42–74; table 1). Interestingly, the His46Arg mutation in ALS141.1 had escaped detection by single stranded conformational polymorphism performed before our study. This patient, who had only one affected sister, was included in this investigation because no information was available on their first or second degree relatives.
Clinical and molecular data of patients with familial amyotrophic lateral sclerosis who have SOD1 mutations
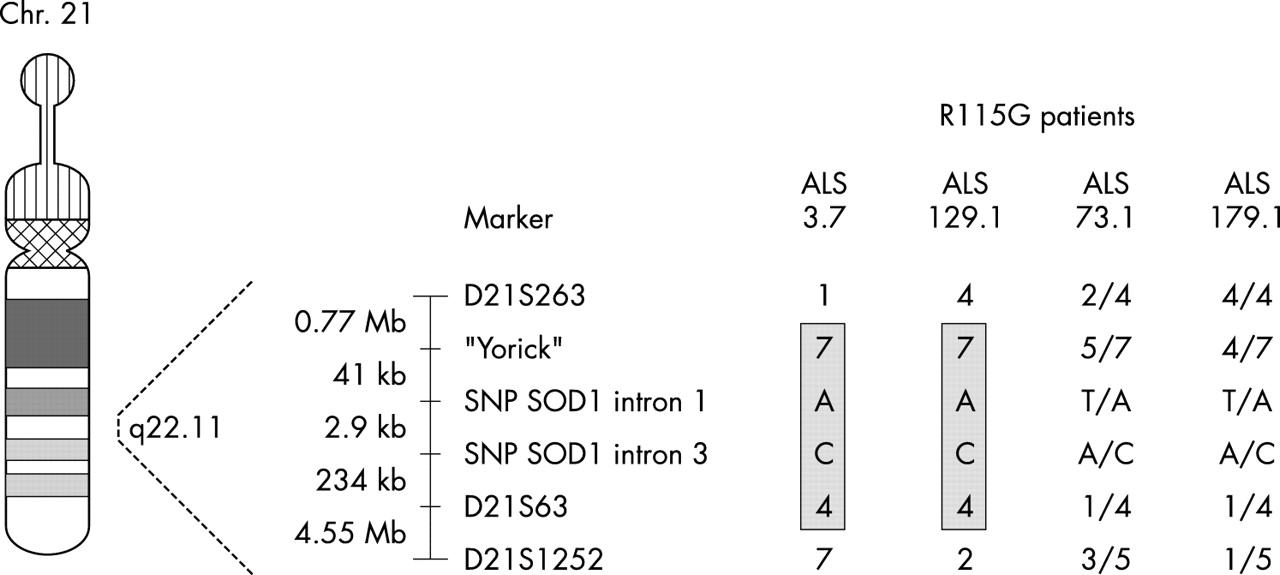
Four (ALS3.7, ALS73.1, ALS129.1, and ALS179.1) of the nine probands carrying SOD1 mutations have the same R115G mutation in exon 4 of the gene. There was no geographical clustering of these four patients’ families. Patients ALS3.7 and ALS129.1 were from Lower Saxony, ALS73.1 was from the Bremen area, and ALS179.1 was from Saxony. Genealogy could be traced for ALS3.7 in three generations, for ALS73.1 in five generations, for ALS129.1 in four generations, and for ALS179.1 in three generations without evidence for relatedness of these families. Moreover, the parental origin of the oldest known generations in the four families revealed no geographical overlap.
Because, regardless of their geographical location, these patients could have derived from a common ancestor, we constructed haplotypes where possible and analysed allele frequencies of polymorphisms (intragenic SNPs and flanking STRPs) at the SOD1 locus on chromosome 21q22. Parental genotypes were available or could be inferred for haplotype construction for two of the four R115G probands (ALS3.7 and AlS129.1). Nondirectiveness of genetic counselling did not allow for blood collection from relatives of ALS73.1 and ALS179.1. As a result, haplotypes could not be constructed in these patients.
A shared haplotype (7-A-C-4) was present in ALS3.7 and in ALS129.1 for four (Yorick, IVS1-108T>A, IVS3+34A>C, and D21S63) of the markers investigated (fig 1). The shared haplotype (7-A-C-4) was not present in the control chromosomes because multilocus genotypes including alleles 7, A, C, and 4 at the respective loci were not observed.
{kind=link}
Disease haplotype and allele sharing in patients with familial amyotrophic lateral sclerosis (ALS) who have the R115G SOD1 mutation. The disease haplotype is shaded. SNP, single nucleotide polymorphism.
Consistent with the haplotype data, there was a significant over-representation of single alleles in the four R115G SOD1 probands. As shown in table 2, the allele “7” of the marker Yorick was present in all four R115G probands and in 21% of the controls. The “A” allele of the SNP in SOD1 intron 1 (IVS1-108T>A) was found in all four R115G probands and in 12% of the controls. Similarly, all four R115G probands carry the “C” allele of the SNP in SOD1 intron 3 (IVS3+34A>C), whereas its frequency in the control population was 10%. The frequency of the “4” allele from the marker D21S63, present in all four R115G probands, was 9% in the control population.
Allelic association of chromosome 21q22 markers in patients with amyotrophic lateral sclerosis who have the SOD1 R115G mutation
DISCUSSION
Our study describes the results of a SOD1 mutation analysis in a large cohort of German patients with FALS. Among the 75 not obviously related patients with FALS, we identified six different mutations in nine probands. Consistent with the literature, the clinical manifestations in these patients and their age at onset were similar to those of patients with sporadic ALS. Four of the nine probands carry the same SOD1 R115G mutation in exon 4 of the gene. The R115G mutation in patient ALS3.7 was the first published SOD1 mutation in German FALS and has, to our knowledge, not been described in another population.10 The predominance of the R115G mutation in SOD1 positive patients in our study suggested that these patients may have derived from a common founder.
Our study demonstrates sharing of alleles from markers at the SOD1 locus in the four R115G probands. Moreover, a common haplotype associated with the disease causing mutation could be constructed in two (ALS3.7 and ALS129.1) of the four patients with the R115G mutation. Genealogical reconstruction of the four R115G families over at least four generations, including the geographical origin, revealed no overlap in ancestry. Taken together with the allele/haplotype sharing our findings strongly suggest a common founder of the R115G mutation, which accounts for a substantial proportion (∼ 44%) of SOD1 associated ALS in German patients of Central European ethnicity. Ancestral genetic founders have also been proposed for other SOD1 mutations (D90A, I113T, and L84F) in different populations, indicating a low frequency of de novo mutations in SOD1.8,11,12
Some SOD1 mutations are present at high frequency in some populations but do not occur in others. For example, the A4V mutation in SOD1 exon 1 has been detected in approximately 50% of American ALS families with mutations in SOD1, whereas it has not been found in other populations.13 Similarly, in our study, the R115G mutation was identified in four of nine patients with ALS carrying SOD1 mutations (44%), but has not been reported to occur in other populations.
A second goal of our investigation was to determine the fraction of FALS cases associated with mutations in SOD1. Few studies4,13,14 have determined the frequency of SOD1 mutations in FALS to confirm the commonly quoted proportion of 20%.15 Most of these studies have relied on single stranded conformational polymorphism, a screening method with a sensitivity less than 100%. We have performed a systematic mutational analysis applying direct sequencing of all SOD1 exons in 75 probands with FALS. To our knowledge, this is one of the largest investigations of SOD1 mutations in FALS in the German speaking Central European population. Our results show that SOD1 mutations are responsible for at least 12% of FALS in the German population.
Acknowledgments
The authors thank the patients for participation in this study.
REFERENCES
Footnotes
-
Competing interest: none declared