Article Text

Abstract
Background Leigh syndrome (LS) is an early-onset progressive neurodegenerative disorder associated with mitochondrial dysfunction. LS is characterised by elevated lactate and pyruvate and bilateral symmetric hyperintense lesions in the basal ganglia, thalamus, brainstem, cerebral white matter or spinal cord on T2-weighted MRI. LS is a genetically heterogeneous disease, and to date mutations in approximately 40 genes related to mitochondrial function have been linked to the disorder.
Methods We investigated a pair of female monozygotic twins diagnosed with LS from consanguineous healthy parents of Indian origin. Their common clinical features included optic atrophy, ophthalmoplegia, spastic paraparesis and mild intellectual disability. High-blood lactate and high-intensity signal in the brainstem on T2-weighted MRI were consistent with a clinical diagnosis of LS. To identify the genetic cause of their condition, we performed whole exome sequencing.
Results We identified a homozygous nonsense mutation in C12orf65 (NM_001143905; c.346delG, p.V116*) in the affected twins. Interestingly, the identical mutation was previously reported in an Indian family with Charcot-Marie Tooth disease type 6, which displayed some overlapping clinical features with the twins.
Conclusions We demonstrate that the identical nonsense mutation in C12orf65 can result in different clinical features, suggesting the involvement of unknown modifiers.
- MITOCHONDRIAL DISORDERS
- NEUROGENETICS
Statistics from Altmetric.com
Introduction
The C12orf65 gene at 12q24.31 encodes a soluble mitochondrial matrix protein belonging to the family of class I peptide chain release factors (RFs) containing a GGQ motif (Gly-Gly-Gln motif) in the translation termination.1 ,2 Mitochondrial translation machinery in eukaryotic cells leads to a biosynthesis of 13 polypeptides that are involved in oxidative phosphorylation.3 ,4 A mitochondrial ribosome has three transfer RNA (tRNA)-binding sites of A (aminoacyl), P (peptidyl) and E (exit) sites which bind to their respective tRNAs on messenger RNAs.5–7 When a mitochondrial stop codon (UAA or UAG) is recognised at the A site of the ribosome, the RFs interact with the peptidyl-transeferase centre of the large ribosomal subunit via its GGQ motif.2 ,8 This conformation triggers the peptidyl-tRNA hydrolysis, which catalyses the hydrolysis of the ester bond between the nascent polypeptide and peptidyl-tRNA.1 ,2 The C12orf65 mutation impairs the oxidative phosphorylation system leading the variable phenotypes like mitochondrial disease.9–14 Knockdown of C12orf65 has been reported to result in mitochondrial translation defect with increased reactive oxygen species production, reduced cellular proliferation and apoptosis.2
In humans, C12orf65 mutations were first described in two unrelated pedigrees affected with Leigh syndrome (LS; MIM #256000) and Leigh-like syndrome.9 C12orf65 mutations produce a variable clinical spectrum with the autosomal recessive inheritance mode. Disease caused by C12orf65 mutations include autosomal recessive spastic paraplegia-55 (SPG55; MIM #615035), Charcot-Marie Tooth type 6 (CMT type6; MIM #601152) and syndromic autosomal recessive intellectual disability.10–14 To date, at least eight mutations in C12orf65 have been reported.9–14 Interestingly, they are all truncating mutations (figure 1). As this gene is known to escape nonsense-mediated decay,9 mutations may produce truncated proteins. It was reported that clinical severity may be dependent on the residual length of the truncated C12orf65 protein and variable sizes of truncated C12orf65 lead to a wide clinical spectrum.

Genetic analysis of the patient. (A) Family pedigree. Black and white symbols show affected and unaffected individuals. One unaffected individual (II-1) was not examined. (B) Electropherograms of II-2 (unaffected) and II-3 (affected). (C) Human C12orf65 protein structure and truncated proteins with previously reported mutations. The orange segment represents a release factor-1 (RF-1) domain. The GGQ motif which has catalytic activity is shown in light blue. The stretch of mutant amino acids resulting from the frameshift is depicted as a red segment. The combination of p.P34Ifs*25 and p.G72Afs*13 was reported as a compound heterozygous mutation by Heidary et al.12 The other eight mutations were identified as homozygous mutations in consanguineous families.
In this paper, we describe a pair of monozygotic twins presenting with LS with a nonsense mutation in C12orf65. Interestingly, the identical mutation was previously reported in patients with Charcot-Marie Tooth type 6, including that differences in clinical presentation can be observed with the same mutation.
Materials and methods
Subjects
We analysed monozygotic female twins with LS (II-3 and II-4), one of their male siblings (II-2) reported of mild muscle pains, and their parents (I-1 and I-2) in this study (figure 1A). Peripheral blood samples were collected after obtaining written informed consent. DNA was extracted from peripheral blood leucocytes using QuickGene-610L (Fujifilm, Tokyo, Japan) according to the manufacturer's instructions. The Institutional Review Board of Yokohama City University School of Medicine approved this study.
Mutation detection
Whole exome sequencing (WES) was performed for one of the affected twins (II-3), as described in the online supplementary methods. On the basis of the autosomal recessive model and the consanguinity in this family, we selected homozygous variants and validated them with the Sanger method. PCR products were sequenced on an ABI3500xL sequencer (Applied Biosystems, Foster City, California, USA) and analysed using Sequencher 5.0 (Gene Codes Corporation, Ann Arbor, Michigan, USA).
Results
Clinical findings
The affected twins (Patients II-3 and II-4 in figure 1A) were 11-year-old girls born to consanguineous healthy parents of Indian-Jewish descent. A 25-year-old brother (II-1) is healthy, and a 15-year-old brother (II-2) has a history of attention-deficit hyperactivity disorder and muscle pain, but muscle biopsy showed no particular abnormality. The pregnancy of the affected twins was remarkable for intrauterine growth restriction (IUGR) noticed since 26 weeks of gestation. The twins were born prematurely at 33 weeks of gestation via cesarean section due to IUGR.
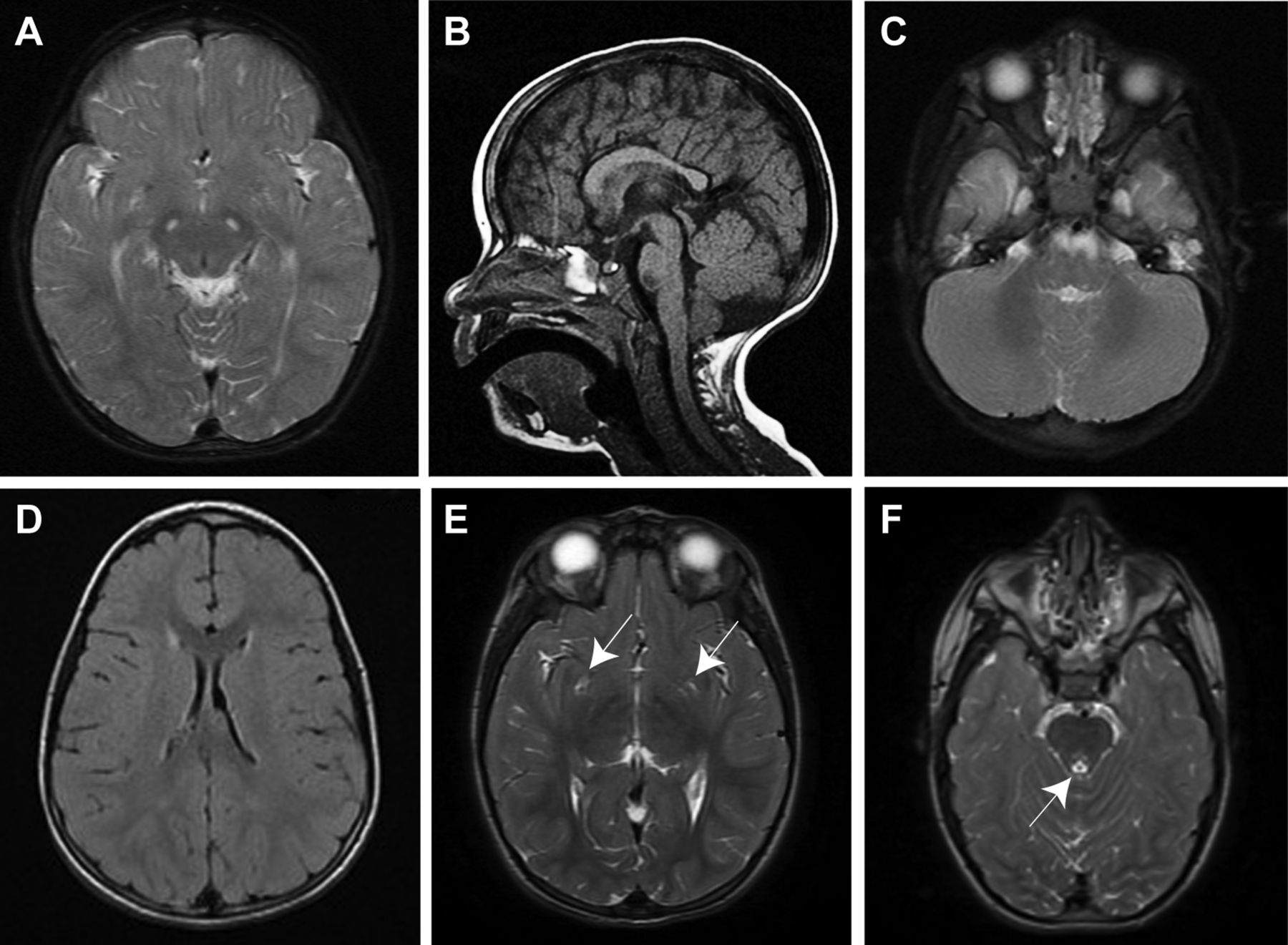
Patient II-3 was born with Apgar scores of 9/10 at 1 and 5 min. Her birth weight was 1320 g (<−3 SD). From approximately 6 months of age, the parents noticed a failure to thrive and developmental delay. Starting at the age of 2 years, she displayed bilateral strabismus, mild horizontal nystagmus, ptosis and ophthalmoplegia. Fundus examination at 2 years revealed temporal optic pallor and optic atrophy. A brain MRI revealed symmetric increased T2 signals in the corticospinal tracts in the brainstem (midbrain and medulla; figure 2A), and the T1-weighted image showed a mildly thick and dysmorphic corpus callosum (figure 2B) while the spine MRI was normal. Serum lactate was markedly elevated (50.9 mg/dL; normal: 6−18 mg/dL) while pyruvate was normal (0.26 mg/dL; normal: 0–1 mg/dL). Urine organic acids showed increased 2-oxoglutaric acid (450 μmol/mmol creatinine; normal: <117 μmol/mmol creatinine) and plasma amino acids were normal. At the age of 2.5 years, she underwent ear tube insertion because of serous otitis media and has normal hearing. During the following years, she exhibited short stature and progressive spastic paraparesis. Physical examination revealed spasticity in the lower extremities with brisk deep tendon reflexes, clonus and extensor plantar response, while the upper extremities were normal in muscle and deep tendon reflexes. Muscle weakness, atrophy and spastic equinovarus were noted only in her lower limbs (see online supplementary video). Superficial and deep sensations were normal in her upper and lower limbs. She started to walk with assistance at 3 years. Currently, she walks on tiptoes with braces and tends to fall frequently. She also fatigues very easily while walking. Growth hormone stimulation test performed at 5 years of age showed a peak growth hormone response of 15.9 ng/mL (normal response: >10 ng/mL). At the age of 5 years, a mild to moderate intellectual disability was recognised and she was referred to a special education school. Currently, she can read and write a few words and perform basic arithmetic. Her left eye esotropia was surgically corrected at 6.5 years. A brain MRI study performed at the age of 7 years revealed hyperintense signals in the bilateral periventricular white matter, bilateral putamen, right side of medial thalamus or periaqueductal area of the midbrain (figure 2D–F). Her fundus was found to show stable temporal pallor at the age of 9 years. At the age of 9.5 years, right hydronephrosis possibly owing to aberrant vessel formation causing ureteral obstruction was found and she underwent pyeloplasty. Currently at 11 years, there are no signs of pubertal development (Tanner stage 1), and she showed markedly elevated follicle-stimulating hormone (FSH) of 45.9 mIU/mL (normal range for age: 0.68−7.26 mIU/mL), borderline luteinizing hormone (LH) of 5.27 mIU/mL (normal range for age 0.02−4.12 mIU/mL) and undetectable oestradiol (E2), consistent with primary ovarian failure. Her physical condition is stable with no regression and no episodes of metabolic decompensation during febrile illness.

{kind=link}
{kind=link}
Brain MRI of affected patients with the C12orf65 mutation. (A, B) T2-weighted (A) and T1-weighted (B) MRI of II-3 showing a symmetric high-intensity signal in the corticospinal tracts at the midbrain level and mildly thick and dysmorphic corpus callosum at 2 years. (C) II-4 at 2 years had an increased signal in the brainstem (pons) on T2-weighted MRI. (D–F) Fluid-attenuated inversion recovery image of II-3 at the age of 7 years showing mildly high-signal intense lesion in the bilateral periventricular area (D), T2-weighted image demonstrating the high signals in the bilateral putamen (arrows), right side of the medial thalamus (E) and the periaqueductal area (arrow) (F).
Patient II-4 is the twin sister of II-3. Her birth weight was 1160 g (<−3 SD) at 33 weeks. She showed a similar clinical course to her twin sister. Brain MRI at 2 years revealed increased T2-weighted signals in the brainstem (figure 2C) and a mildly dysmorphic corpus callosum, while the MRI of the spine was normal. At 3 years, high serum lactate and normal pyruvate were found (56.2 mg/dL and 0.18 mg/dL, respectively). Growth hormone stimulation test performed at 5 years of age showed a peak growth hormone response of 10 ng/mL. She underwent adenoidectomy at the age of 6 years because of adenoidal hypertrophy. She was operated for Achilles tendon lengthening at 9.5 years and is using braces as well. She had a urinary tract infection recently, but her kidney ultrasound revealed no renal abnormalities unlike her sister. At 11 years, she is at Tanner stage 1 and was also diagnosed as primary ovarian failure with elevated FSH (73.7 mIU/mL) and elevated LH (23.9 mIU/mL) and undetectable E2.
Muscle biopsy at the age of 4 years revealed decreased complex IV activity (cytochrome c oxidase: 750 nmol/min/mg and 770 nmol/min/mg in II-3 and II-4, respectively, normal range: 1413±518 nmol/min/mg), while the other mitochondrial complexes (I, II, III and V) were normal (see online supplementary methods and table S1). Both patients were suspected to have LS due to the clinical manifestations and symmetric lesions of brain MRI at 2 years, and finally diagnosed as LS at 4 years by the decreased complex IV activity. They were treated with growth hormones between the ages of 6 and 8 years, but there was no response (see online supplementary figures S1 and S2). They were also treated with coenzyme Q10 between the ages of 3 and 7 years and with creatine monohydrate between 9 and 10 years, but without remarkable effect. Both karyotypes were found to be of a normal female individual.
Identification of the pathogenic mutation
In WES analysis of patient II-3, 95.2% of coding sequences were covered by at least 20 reads. After in silico analysis, 24 homozygous candidate variants were extracted (see online supplementary table S2). Among these was a homozygous nonsense mutation in C12orf65 (c.346delG, p.V116*; NM_001143905), a gene in which mutations have been previously reported in LS. Sanger sequencing revealed a homozygous change in the affected twins and a heterozygous change in their parents, consistent with the autosomal recessive inheritance pattern. Their elder brother (II-2) did not have this mutation (figure 1B). The identified mutation was absent from the 1000 Genomes database, NHLBI Exome Sequencing Project (ESP6500) and our in-house exome database (n=982). No pathological variants in five nuclear genes (SURF1, COX15, SCO2, TACO1 and LRPPRC) and mitochondrial DNA, which have been reported in relation to LS with COX deficiency, were identified (see online supplementary table S3 and figure S3).
Discussion
C12orf65-related diseases are characterised by a triad of core phenotypes including visual difficulty (optic atrophy and ophthalmoplegia), pyramidal signs (spasticity, spastic paraplegia and pathogenic reflexes) and peripheral neuropathy, as well as variable cognitive or intellectual impairment.9–14 In previous reports, all of the C12orf65 mutations were truncating, including four frameshift mutations (p.P34Ifs*25, p.G72Afs*13, p.V83Gfs*2 and p.K138Rfs*17), three nonsense mutations (p.V116*, p.R132* and p.R139*) and one splicing mutation (skipping of exon 2; see online supplementary table S4).9–14 Patients with truncating mutations close to the C-terminal (p.R132*, p.R139* and p.K138Rfs*17) showed only core phenotypes; they displayed mild intellectual disability and almost normal cognitive ability, but no abnormal brain lesions.10 ,11 ,13 In contrast, the patients with truncations terminating close to the N-terminal (p.P34Ifs*25, p.G72Afs*13 and p.V83Gfs*2) exhibited severe phenotypes, including neonatal death, bulbar dysfunction, severe cognitive impairment and intellectual disability with high-intensity lesions in the brainstem, pons, thalami and midbrain in T2-weighted MRI, in addition to the core phenotypes.9 ,12 Thus, the phenotypic severity appears to depend on the length of the truncated C12orf65 protein.13
Here, we found a homozygous nonsense mutation in C12orf65 (p.V116*) in female LS twins with optic atrophy, ophthalmoplegia, short stature, mild intellectual disability, spastic paraparesis, high lactate and symmetric hyperintense lesions in T2-weighted brain MRI. The mutation results in a mid-length truncation (figure 1C), and the clinical features are moderate. This is consistent with the concept that the extent of the truncation correlates with disease severity. The identical p.V116* homozygous mutation was previously reported in CMT type 6 by Tucci et al (figure 1C).14 Interestingly, the patients with CMT type 6 and the presently affected twins have Indian ancestry; thus, there is a possibility that the p.V116* mutation might originate from the same founder. However, the present LS cases exhibited a more severe clinical phenotype compared with the patients reported by Tucci et al, and the clinical features within each family are quite unique. Furthermore, the present twins displayed decreased mitochondrial complex IV activity, while the patients with CMT type 6 showed decreased complex V activity (without no information of I-IV complex activities; table 1). These differences in clinical presentation and mitochondrial complex activities suggest that additional genetic factors such as unidentified mutations/modifiers by WES or genetic background including common variants may contribute to such phenotypic variability in patients with the identical C12orf65 mutation. WES may also miss variants at regulatory regions.
Clinical features of the present patients and previously published cases with the p.116* homozygous mutation
In conclusion, we identified the C12orf65 nonsense mutation (p.V116*) in the affected female twins with LS. The length of the truncated protein appears to inversely relate to the severity of the phenotype. However, further studies on C12orf65 mutations are required to clarify the phenotype-genotype correlations.
Acknowledgments
The authors thank the patient and her family for participating in this study. The authors also thank Ms S Sugimoto and K Takabe for their technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online video
Footnotes
EI and AF-V contributed equally to this study.
Contributors EI performed the experiments, interpreted the data and wrote the manuscript. AF-V recruited the patients, performed the clinical evaluation and wrote the manuscript. OE performed endocrinological evaluation and wrote the manuscript. AS performed and evaluated the mitochondrial enzyme assay. SM, MN, YT and HS contributed to data analysis. NMI and NMA conducted and supervised this study, evaluated the data and wrote the manuscript.
Funding This work was supported by research grants from the Ministry of Health, Labour, and Welfare of Japan (H S, N Miyake, N Matsumoto; 24118007, 25293085, 25293235, 26670505), the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Ministry of Education, Culture, Sports, Science and Technology (N Matsumoto), the Strategic Research Program for Brain Sciences (N Matsumoto; 11105137), a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (N Miyake, N Matsumoto; 12024421), and the Takeda Science Foundation (N Miyake, N Matsumoto).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethics approval for this project was obtained by the Institutional Review Board of Yokohama City University School of Medicine.
Provenance and peer review Not commissioned; externally peer reviewed.