Article Text

Abstract
OBJECTIVES To describe the consequences of the identification of the Huntington's disease (HD) mutation on predictive and prenatal testing.
METHODS A retrospective study was performed considering the test applicants, procedures, and results before and after the identification of the mutation. 1032 people at risk for Huntington's disease in The Netherlands were included, of whom 741 applied for the predictive test in the period 1987 to 1997 in Leiden at the Department of Clinical Genetics, and after 1994, also in the other seven clinical genetics departments in The Netherlands. Uptake, sociodemographic variables, and test results, taken before and after the mutation was identified, are described.
RESULTS The uptake of the predictive test in the period studied was 24% and for the prenatal test 2%. No differences were noted in numbers and sociodemographic data between the period before and after the mutation was identified. After an initial increase in test applicants, a decrease was seen after 1995. After 1993 a significant increase of 25% at risk test applicants and a significant decrease of prenatal exclusion tests was noticed. Only 7% asked for reassessment by mutation analysis. New problems arose after the identification of the mutation, such as the option of reassessing the risk obtained by linkage analysis, direct mutation testing of 25% at risk persons with a parent who does not wish to know, new choices regarding reproduction, and new uncertainties for carriers of intermediate and reduced penetrance alleles and for their offspring and relatives.
CONCLUSIONS Although predictive testing has become reliable and available for every person at risk since the mutation has been identified, the uptake of predictive and prenatal tests fell short of expectation, no change in sociodemographic variables was seen, and a decrease in number of applicants was noted. Furthermore, new uncertainties, psychological problems, and questions arose.
- Huntington's disease
- predictive testing
- prenatal testing
- mutation analysis
Statistics from Altmetric.com
Huntington's disease (HD) is an autosomal dominant, progressive neuropsychiatric disorder. Because the mean age at onset is 40 years, the risk of a person with an affected parent is still about 50%, when decisions about future and family planning are being made in early adult life.1 After localisation of the Huntington's disease gene to chromosome band 4p16.3 in 1983, predictive testing using linkage analysis became available.2 When the mutation was identified in 1993,3 testing became technically more simple, reliable, and available for every person at risk.
Before the predictive test became available, 57% to 84% of the at risk persons indicated interest in the predictive test.4-6 Since its introduction in 1987, the uptake of only 2%–16% fell short of expectations.7-9 Reasons for taking the test were, reportedly, to end uncertainty, to obtain control over the future, and to inform offspring and relatives. Reasons for not taking the test were predominantly listed as concern for the possible increased risk of children, the absence of treatment, the potential loss of health insurance, the financial costs of testing, and the inability to undo the knowledge. The main reason, however, seemed to be the fear of being unable to cope when receiving a high risk result.4 8 10-12 Carriers were reported to show relief from psychological distress and a tendency to minimise the impact of the test. A substantial number of non-carriers experienced no relief, numbed emotions, survivor's guilt, and suffered in developing a new life perspective.10 13 14 Catastrophic events have only been seen occasionally.15
We presumed that the direct test for Huntington's disease would mean a better test and would therefore attract more at risk persons for predictive and prenatal testing and solve linkage related testing problems. The aim of this study was to examine the consequences of the identification of the mutation on predictive and prenatal testing for Huntington's disease.
Subjects and methods
In The Netherlands the estimated prevalence of Huntington's disease is 6.5:100 000, based on the number of living affected persons, recorded at the Leiden roster for Huntington's disease. Together, at least 3115 persons at 50% risk are registered.
Predictive testing for Huntington's disease was centralised at the Department of Clinical Genetics in Leiden from October 1987 until 1994. In 1994 the other seven departments of clinical genetics in The Netherlands started genetic counselling for Huntington's disease.
The applicants described in this study were those counselled in Leiden between October 1987 and December 1997. The Dutch Working Group on Huntington's disease provided the total number of predictive tests in The Netherlands.
A structured predictive testing procedure for Huntington's disease was introduced in 1987 in Leiden10 and the testing protocol, following the international guidelines,16 17 includes at least two pretest and one post-test counselling session. Psychological support is offered in the decision making process, before and after disclosure.
DNA analysis for Huntington's disease in The Netherlands remained centralised in Leiden. Before 1993, testing was performed by linkage analysis. Once the mutation was identified, the status of the at risk applicant could be assessed by direct mutation analysis. In each family DNA from an affected relative was analysed to confirm that this mutation was the cause of Huntington's disease in the family.
Differences between groups were analysed using Student'st test, Fisher's exact test, or χ2 tests, and p=0.05 was set as the criterion for significance.
Results
The total number of persons applying for predictive testing for Huntington's disease in The Netherlands in the period 1987 to 1997 was 1032. Of these, 752 (73%) decided to be tested (table 1)—that is, 24% of the at risk persons registered in the Leiden Roster for Huntington's disease. The total number of test applicants increased up to 1995 and decreased thereafter. After an initial increase in numbers up to 1994, the number of predictive test applicants/year in Leiden decreased (fig 1).
Total number of applicants, and numbers of applicants tested and not tested in Leiden (n(%)) specifically, and in The Netherlands in general (1987–97)
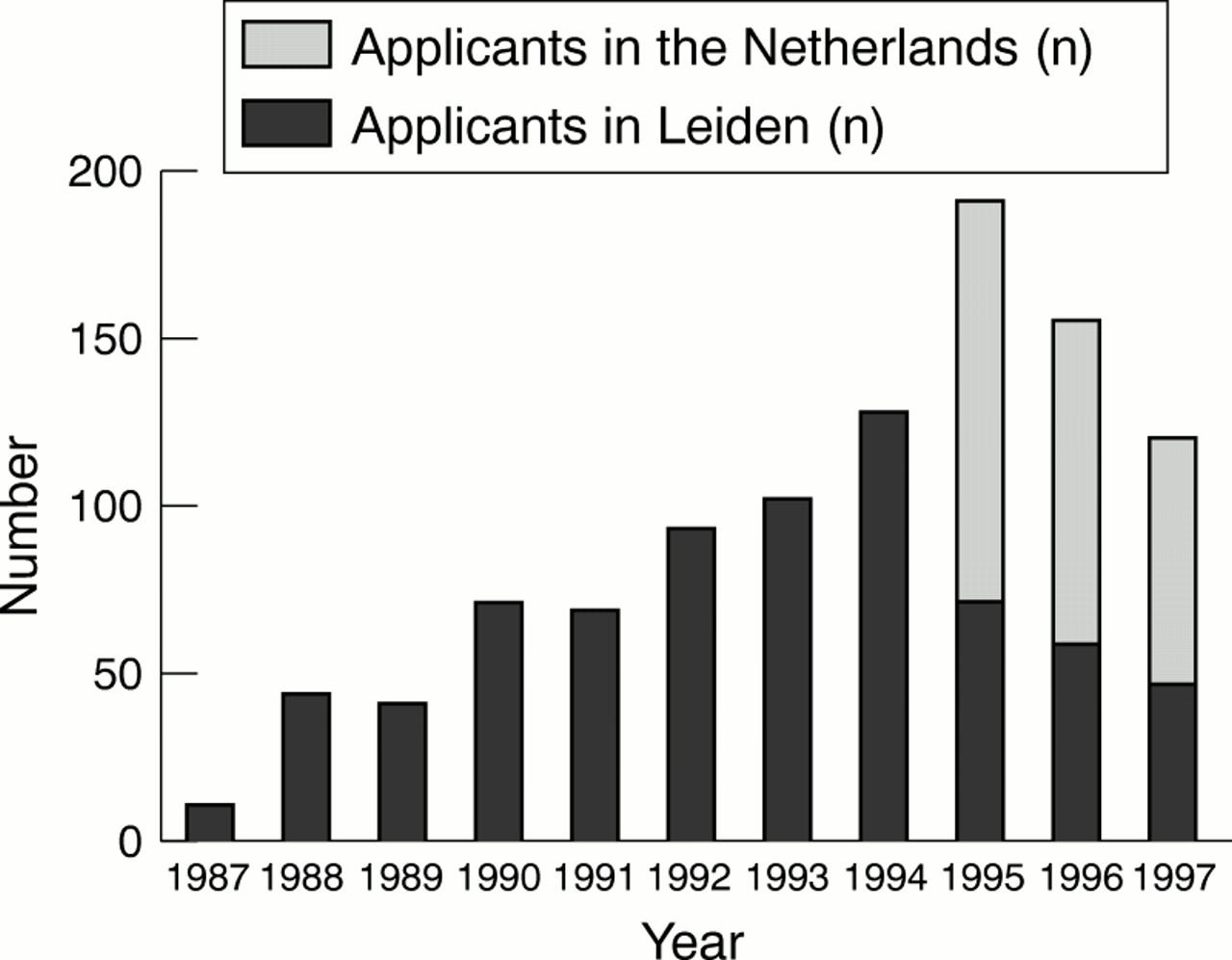
Number of persons applying for predictive testing for Huntington's disease in Leiden and The Netherlands/year (1987–97).
The following results are only from the Leiden data: 741 at risk persons have applied for predictive testing. Seventy seven per cent (n=570) of these applicants decided to be tested (table 1) resulting in an increased risk for 41% (n=234). Fifteen per cent (n=112) withdrew from testing, 2.5% (n=19) decided not to be tested themselves but asked for a prenatal exclusion test, and 2% (n=17) complained of symptoms, and were referred to the Department of Neurology. Due to linkage analysis problems a definitive result was not obtained because of an uninformative test result (n=12;1.5%), rest risk (242 tests with a mean reliability of 97.2% (range of rest risk:1–9%)), and 3% (n=23) could not be tested because their family structure was limited.
The 35 applicants who could not be tested because of their family structure or uninformative results, were informed by our Department about the new test in 1993. Almost all (92%) who received an uninformative test result and 30% who could not be tested because of their family structure decided to be tested by mutation analysis. Seven of the 242 tested were retested because of prenatal diagnosis and 11 because of symptoms by the neurologist. Only 16 of 224 test applicants (7%) requested reassessment themselves because they wanted the most reliable test.
The sociodemographic data of predictive test applicants from Leiden are given in figure 2 A and B. The mean age (35 years), percentage having children (44%), and sex ratio (M:F(%)=41:59) did not vary significantly in time (fig 2 A). The relative number of carriers (41%) and applicants not tested (23%) fluctuated but also remained more or less stable over time (fig 2 B). There were no significant differences between age, sex ratio (p=0.09), or having children (p=0.81) between those tested or not tested, and before or after the mutation was identified.
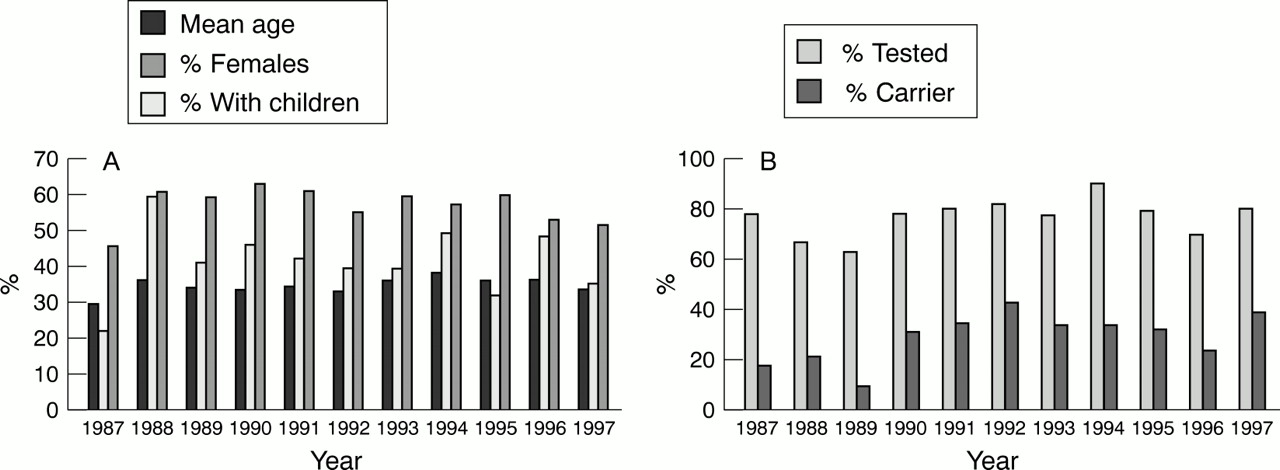
(A) Sociodemographic data (mean age, relative number with children, relative number of females) of persons applying for predictive testing for Huntington's disease in Leiden, The Netherlands/year (1987–97). (B) Relative numbers tested and relative number of carriers in persons applying for predictive testing for Huntington's disease in Leiden, The Netherlands/year (1987–97).
Before the mutation was identified 42% of the families versus 53% after, had only one applicant (p=0.07).
After the mutation was identified, about one third of the requests for predictive testing came from applicants from families in which the diagnosis had become recently known by mutation testing of an affected relative.
The age distribution and the mean age of men and women were comparable (fig 3). Most applicants applying for predictive testing (56%) had no children, were younger than 40 years, and of both sexes (table2).

Age and sex distribution of predictive test applicants for Huntington's disease in Leiden, The Netherlands (1987–97).
Test applicants for Huntington's disease sorted by sex, parental status, and age in Leiden, The Netherlands (1987–97)
Most of the applicants (n=676; 90%) had a prior risk of 50%, whereas 65 (10%) had a prior risk of 25%. The mean number of test applicants at 25% risk rose significantly from 3.7/year to 9.0/year after the mutation was identified (p<0.001; χ2=12.51, df=1; fig4). The most frequent choice before the mutation was identified was testing the at risk parent first (38%) and after the mutation test was available (41%). The exclusion-definitive test was chosen by 18%. Before the mutation was identified, 33% of the 25% at risk applicants could not be tested, and after the mutation, 15% withdrew from testing. Unsought information due to predictive testing, which means either a change in prior risk or being a carrier or not, was obtained for 0/12 parents and 9/15 sibships before, and 3/23 parents and 12/30 sibships after the mutation was identified. The mutation test caused significantly less unsought information than the exclusion-definitive test (8% v 100%; Fisher's exact test, p=0.033). According to the follow up of the test applicants the unsought information did not lead to major problems.18

Number of 25% at risk predictive test applicants for Huntington's disease/year in Leiden and The Netherlands (1987–97).
Once the mutation was identified sporadic patients could be tested. We counselled 12 relatives (2% of the test applicants) from four families. Two of the 12 applicants decided not to be tested, four showed normal results, and six were carriers of an intermediate allele. These applicants were, compared with the other test applicants, older (mean age 44 years) and most of them (75%) had children (p=0.030) at the time the diagnosis was established.
We have performed 31 prenatal exclusion tests (43%) and 41 prenatal direct tests (57%) in 43 couples at risk. This resulted in termination of 28 pregnancies (39%) with an increased risk. In 28 couples (65%), the woman was at risk. The relative number of exclusion tests compared with direct tests has diminished since the mutation was identified (fig5). The prenatal exclusion-definitive test was rarely used (3%). For at least 2% of the persons at risk younger than 50 years, 11% of the tested at risk persons, and a small group of at risk persons wishing not to be tested themselves, especially women, prenatal testing for Huntington's disease seemed an acceptable choice regarding reproduction.19

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Prenatal exclusion and direct tests for Huntington's disease in The Netherlands (1987–97).
Discussion
The number of predictive tests/year in Leiden increased until 1994 and decreased thereafter (fig 1). This can be explained by the decentralisation of the counselling. However, the number of predictive tests in The Netherlands also decreased after 1995, probably reflecting the clearing of a build up of people who had been waiting for the mutation test. Before the mutation was identified fewer single members of the registered Huntington's disease families applied than after. A stimulating effect could be imagined when family members were involved in predictive testing, as it was in the linkage era. After the mutation was identified, a third of the predictive test applicants were persons with a recent family diagnosis. This might be due to the fact that the demand among the known at risk persons is diminishing and that the mutation test provides new (diagnostic) testing possibilities.
The percentage of those at risk tested in The Netherlands (24%) based on the number of registered at risk persons, is significantly higher (95% confidence interval: 22.6–25.6%) compared with the numbers described in the literature (2%-16%).7-9 An explanation might be that in a small country like The Netherlands, which had centralised Huntington's disease counselling in Leiden from 1987 until 1994 and after 1994 annual meetings of the Working Group on Huntington's disease, where DNA analysis for Huntington's disease had also been centralised in one laboratory and where an extended roster for Huntington's disease exists, we were able to get an accurate view of the uptake. It might, however, be that the number of at risk persons is underestimated as there may be people not included. The percentage at risk persons tested in The Netherlands, based on the number of first degree relatives according to Conneally (five times the number of affected persons)20 and on the mean European prevalence (4:100 000),1 is however 25%, not significantly different from the 24%, previously mentioned.
It is remarkable that the relative number of persons tested per year remained constant in time, (fig 2 B), because when only linkage analysis was available some could not be tested or provided with a test result, due to problems such as non-paternity, recombination, unavailability of family material, or an uninformative family structure. Furthermore, the mean age and the relative number of applicants having children did not decrease (fig 2 A) although genetic counselling and predictive testing has become more acceptable and accepted through public education and is now available for every person at risk. Perhaps for most, predictive testing is not an option to improve the quality of life. Alternatively, more time may be needed—for example, the time of one generation—to show an effect.
As described in earlier reports more female (58%) than male (42%) persons at risk applied for predictive testing; likewise, more women at risk (65%) applied for prenatal testing in our series. This difference might reflect greater maternal involvement in reproductive decisions or concern for existing children at risk. Men may experience greater difficulty in accepting the implications of being at risk and are less able or willing to deal with the emotions that risk alterations arouse, coping by denial rather than confronting the issue.8Another explanation might be that men choose other reproductive options.19
The age distribution and mean age of male and female applicants are comparable (fig 3).
Most test applicants of either sex did not have children and were younger than 40 (table 2). This is in accordance with the notion that family planning is one of the main reasons to be tested.
Significantly more applicants in this study, 23% (95% confidence interval 20–26.1%), decided not to be tested as opposed to 18% described in other studies.12 The sociodemographic variables of those tested and not tested were comparable and no decrease in number of those not tested, their age, or their parental status, in time was seen.
That more people received a decreased (59%) than an increased risk (41%) can be explained by the age distribution (mean age 35 years, age adjusted risk for a person at 50% risk is 45.5%21) and due to a lower prior risk of about 10% of applicants. Furthermore, other factors may play a part such as self selection,11some at risk persons perceive that they are free of symptoms, becoming evident in (younger) siblings, and are thus encouraged to seek predictive testing in the expectation of a favourable result. Alternatively, cognitive changes associated with Huntington's disease might reduce the motivation to be tested.7
Once the mutation was identified some technical testing problems were solved, but the following new dilemmas emerged:
The choice of reassessment of the risk obtained by linkage analysis—Reassessment analysis was, as in other countries,22 not often performed, although the range of the residual risk was 1% to 9%. This might be due to the fact that the people either accept to live with this residual risk, are frightened to be found to be a carrier after all, are not informed, do not understand the implications of the test result, or deny the rest risk.
New testing options for predictive testing of 25% at risk persons with a 50% at risk parent who does not wish to know his/her genetic status—that is, the exclusion-definitive test and the mutation test. The outcome of these new options does not only disclose the risk of the test candidate, but may also change the risk of the at risk parent and siblings.18 Before the mutation was identified this was only possible by the exclusion test, ensuring the parent's wish not to know.
New choices regarding reproduction—These emerged after the mutation was identified, but the uptake for prenatal testing has remained low.5 18 For 2% of the at risk persons younger than 50 years—that is, 11% of the tested carriers and a small group of at risk persons wishing not to be tested themselves—prenatal testing seems an acceptable choice regarding reproduction. Although prenatal testing became relatively simple, reliable, accessible, and more acceptable and although family planning is one of the main reasons why predictive testing is requested, only a few people at risk make use of the service. This can partly be explained by the mean age of the predictive test applicant (35 years) and the fact that 44% already had children.19 The low uptake is probably related to the wish not to be informed about their own genetic status, the fear of being unable to cope with an unfavourable test result, termination, or risk of the prenatal test. Furthermore it might be explained by psychological defence mechanisms, such as denial and minimalisation,10 or the ethical reservations about termination of pregnancy for a late onset disorder or the hope that a therapy might become available.19Nowadays, direct mutation testing of the fetus alone is technically more simple and fast although the exclusion-definitive test and the direct mutation test of the fetus alone both have the dilemma of disclosure of the parental genetic status simultaneously with that of the fetus with a probable termination of pregnancy, and the risk of disclosure of the genetic status of the parent is smaller. This test should therefore be offered as an option and included in the international guidelines.19
New and often unexpected results of predictive testing, new uncertainties about the risk for Huntington's disease, specific psychological problems, and new questions—These arose for carriers of intermediate alleles or alleles with reduced penetrance and their offspring, which made counselling and choices for those at risk difficult. New uncertainties concern the lack of reliable penetrance risks and risks for expansion of 27–39 repeats. Alleles of 27–35 CAG repeats have demonstrated meiotic instability in sperm, increasing with more CAG repeats but have not been associated with a Huntington's disease phenotype so far.23 24 The likelihood that transmission of an allele in this range will result in a Huntington's disease allele is unknown, but depends on the sex of the transmitting person, the size of the CAG repeat, and possibly unknown factors. Although 36–39 CAG repeats have been clinically and pathologically associated with a Huntington's disease phenotype it is not always penetrant in these people.24 Specific psychological problems were seen in those with 27–35 repeats who decided to be tested in the hope of relieving their children of the problem. Although receiving a decreased risk to develop the disease themselves, their children are still at risk, which may result in feelings of guilt in the parents and reproach in the children. Other psychological problems concern risk uncertainty and the difference of either test result for sons and daughters. Informing a family, unaware of being at risk, about the uncertain consequences of a repeat size between 27 and 39 and offering testing may also induce much turmoil. New questions are being considered about how should newly found carriers of 27–39 repeats be approached and counselled and should relatives be informed, to whom should predictive and prenatal testing be offered, and which fetal repeat size is acceptable to terminate a pregnancy?
The primary objective of predictive testing is to support and help people make decisions based on their own judgement and in their own interest, and to provide options that may improve the quality of their lives. Although, paradoxically, testing has become technically easier, more reliable, and available for every person at risk now the mutation is identified and the predictive test has been shown to have potential benefits,14 only a small, self selected group of at risk people applied for predictive testing. Most of the at risk persons did not consider the test an option to improve the quality of their lives; the improved test did not convince them otherwise.
Acknowledgments
We thank Professor M Breuning for critical reading of the manuscript, Dr H van den Boer-van den Berg for her critical and stimulating remarks, R Belfroid for performing DNA analysis, the Dutch Working Group on Huntington's disease for providing the total numbers of predictive and prenatal tests, Dr R Giles for language editing, and Dr A Zwinderman for statistical analysis.