Article Text

Abstract
OBJECTIVES To define the molecular genetic basis of the MELAS phenotype in five patients without any known mutation of mitochondrial DNA.
METHODS Systematic automated mitochondrial DNA sequencing of all mitochondrial transfer RNA and cytochrome c oxidase genes was undertaken in five patients who had the MELAS phenotype.
RESULTS A novel heteroplasmic mitochondrial DNA mutation was identified in the transfer RNA gene for phenylalanine in one case (patient 3). This mutation was not detected in the patient’s blood or in her mother’s blood. No pathogenic mutations were identified in the other four patients.
CONCLUSIONS This is the first point mutation in the transfer RNA gene for phenylalanine to be associated with MELAS. The absence of mutations in the remaining four patients suggests that there is further genetic heterogeneity associated with this mitochondrial phenotype.
- MELAS
- mitochondrial DNA
- tRNA gene
Statistics from Altmetric.com
The first description of a disorder which included stroke-like episodes, lactic acidaemia, and ragged red fibres was by Shapiraet al in 1975.1 Pavlakis et al 2 described further cases, introduced the acronym MELAS (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes), and suggested that this represented a distinct mitochondrial disease phenotype. Subsequent reports have established the central role of mitochondrial respiratory chain dysfunction in the pathogenesis of this disorder.3 4 In 1990 Goto et al. 5 identified a point mutation in the transfer RNA (tRNA) leucine(UUR) gene in some patients with MELAS. This has now been established to be the commonest mitochondrial DNA (mtDNA) defect described in association with MELAS although it is not specific to the MELAS phenotype.6 Subsequent studies have identified additional point mutations indicating that MELAS is a genetically heterogeneous disorder.7 However, there remains a group of patients with typical stroke-like episodes, ragged red fibres on muscle biopsy, and lactic acidaemia who do not harbour any of the known defects of mitochondrial DNA. We have analysed mtDNA in five such patients and in one of them have identified a novel heteroplasmic mtDNA tRNA gene point mutation.
Patients and methods
PATIENT 1
A 30 year old woman was a normal full term delivery and had normal motor and cognitive milestones. At the age of 23 years, over the course of a few days, she developed a left hemiparesis headache, nausea, and vomiting culminating in a generalised seizure. Examination showed a left homonymous hemianopia and mild left sided pyramidal weakness. A right occipitotemporal high signal area was seen on T2 weighted MRI consistent with an area of infarction but not conforming to a single vascular territory. Plasma lactate was raised at 4.1 mmol/l (normal <1.6). Nerve conduction studies showed changes consistent with a subclinical sensory axonal neuropathy but EMG was normal. Muscle biopsy showed that 10% of fibres were ragged red with the succinate dehydrogenase (SDH) stain. All the ragged red fibres (RRFs) were cytochrome c oxidase (COX) positive. The patient had no further stroke-like episodes but developed epilepsia partialis continua of her left arm resistant to treatment with anticonvulsant drugs. Examination at the age of 30 years showed a left homonymous hemianopia and some limitation of eye movements in all directions consistent with a mild external ophthalmoplegia, but with no ptosis. There was horizontal gaze evoked nystagmus in both directions of gaze and she had a persistent no-no head tremor. The left hand was held flexed at the wrist and there was some clawing. There was rhythmic flexion or extension jerking of the left wrist which on EEG back averaging was shown to have a clear cortical correlate. There was no limb weakness and the rest of the examination was normal. There was no family history of neurological disease.
PATIENT 2
This left handed man developed sensorineural deafness in childhood but was otherwise well until the age of 52 when over the course of a few days he developed headaches, nausea, vomiting, and an unsteady gait with a tendency to veer to the left. This culminated in left focal motor seizures affecting his arm and leg. Examination showed a mild left hemiparesis. Brain CT showed a low density area in the right parietal lobe consistent with an area of infarction. He made an initial recovery then represented three weeks later with a sudden increase in weakness of his left side followed by left focal motor seizures. He became confused and drowsy and an EEG was consistent with non-convulsive epileptic status. Plasma lactate was raised at 7.49 mmol/l. T2 weighted brain MRI showed high signal bilaterally in the occipital regions and in the right temperoparietal region. Although he improved after this second stroke-like episode he had a fixed neurological defecit comprising fluent dysphasia, cortical blindness, and a dense left hemiparesis. Muscle biopsy showed 10% ragged red fibres with the SDH stain which were all COX positive. He died 6 months after the initial presentation from bronchopneumonia. There was no family history of neurological disease.
PATIENT 3
A 32 year old woman developed normally until the age of 12 years when she had an acute episode of headache, photophobia, vomiting, and left arm focal motor fitting from which she fully recovered. She experienced two further similar episodes and after the second developed a dense left hemiplegia which subsequently resolved. There followed progressive cognitive decline, cerebellar ataxia, and complex partial seizures. Clinical examination when aged 29 disclosed a salt and pepper type retinopathy, cerebellar ataxia, mild spasticity in all four limbs, and intermittent dystonic posturing of her upper limbs as she walked. There was no limb weakness. Brain CT showed extensive low density areas involving both grey and white matter, most marked posteriorly. These were consistent with areas of infarction. Plasma lactate was raised at 4.9 mmol/l. Muscle biopsy when aged 12 years showed 3% ragged red fibres with the SDH stain which were all COX positive. There was no family history of neurological disease.
PATIENT 4
This right handed man was well until the age of 7 years when he developed episodic headaches with nausea, vomiting, and photophobia which were diagnosed to be common migraine. At the age of 9 years, in addition to the headaches he was having difficulty reading small print and was found to have bilateral optic atrophy. His headaches settled and he was otherwise well until the age of 34. At this age he became confused over the course of a day and was found to have a right homonymous hemianopia, non-fluent dysphasia, and a mild right hemiparesis. He made a complete recovery but at the age of 46 developed sudden onset bilateral visual loss with visual acuities reduced to 6/60 in both eyes, and confusion. Brain scan showed areas of decreased attenuation in both occipital lobes consistent with areas of infarction. He subsequently developed generalised seizures and progressive cognitive decline. Examination at the age of 50 showed marked cognitive impairment with disorientation in time and place. He had bilateral optic atrophy and a salt and pepper type retinopathy. He had a broad based gait with limb incoordination and he exhibited generalised dystonic posturing. There was no clinical evidence of a myopathy. He had bilateral grasp reflexes and generalised hyperreflexia. Plasma lactate was raised at 4.4 mmol/l. Muscle biopsy showed 30% ragged red fibres with the SDH stain which were all COX positive. Polarography of freshly isolated muscle mitochondria showed a defect of complex I. There was no family history of neurological disease.
PATIENT 5
This right handed man had normal motor and cognitive milestones but developed sensorineural deafness in his late teens. He developed cataracts in his 30s requiring surgery. At the age of 45 he experienced headaches, vomiting, and increasing confusion over a period of three days culminating in a generalised seizure. Examination disclosed a fluent dysphasia, dyslexia, dysgraphia, and a right homonymous hemianopia. Brain CT disclosed a low density area in the left occipitoparietal region. He recovered fully but continued to have generalised seizures. At the age of 53 years he had a respiratory arrest after a generalised seizure and was ventilated. His conscious level remained impaired despite the absence of sedation. Examination showed roving eye movements and generalised stimulus sensitive myoclonus. His best motor response was flexion to pain. He had frequent focal motor seizures affecting his face and arms. Plasma and CSF lactate were raised at 8 and 7 mmol/l respectively. Brain MRI showed generalised atrophy and high signal areas in the paramedian regions extending from the medulla up to the medial thalamus, the appearance being consistent with Leigh’s disease. Muscle biopsy showed 10% ragged red fibres with the SDH stain. Most of the ragged red fibres had increased COX staining and there were no COX negative fibres. He was successfully removed from ventilation but his conscious level remained unchanged. He died 2 months later after a further respiratory arrest. There was no family history of neurological disease.
METHODS
The clinical details and results of mtDNA sequencing of the patients studied are summarised in tables 1 and 2. All initial sequencing and Southern blotting were performed on mtDNA extracted from muscle as this tissue is more likely to contain higher amounts of any mutation identified.6 When a change was identified in muscle, mtDNA from blood would then be analysed. Total DNA was extracted from muscle and blood and Southern blotting was performed using methods previously described.17 18 Systematic sequencing of all mitochondrial transfer RNA and cytochrome c oxidase genes was undertaken using a Taq FS dye primer cycle sequencing kit and an automated DNA sequencer (Applied Biosystems Incorporated). The oligonucleotide primers used to amplify each region of mtDNA of interest were tagged with the M13 sequence at their 5′ end. The primer sequences are available from the authors on request. The presence of the G583A point mutation was confirmed by a mismatch polymerase chain reacton (PCR) technique in which a restriction site for the endonuclease Cla-1 was introduced in the presence of the mutant nucleotide. The primers used for the mismatch PCR were, light strand: 371–390, and heavy strand: 603–584 TGCTTTGAGGAGGTAATCGA (mismatch nucleotides are shown in bold). This primer pair produce a 233 bp product. This product is only cleaved into two fragments of 216 and 17bp if the mutant nucleotide is present. The conditions for the mismatch PCR were as follows: one cycle of 94° for 3 minutes, followed by 30 cycles of 92° for 30s, 55° for 30s, and 72° for 30 s, and finally one cycle of 72° for 10 min. Quantitation of the proportion of mutant mtDNA in the muscle of patient 3 was undertaken using a fluorescent method. Fluorescently labelled deoxynucleotide triphosphate was added to the last cycle of the mismatch PCR followed by digestion of the products with Cla-1. The products were then quantified using a 373 DNA sequencer and Gene-scan software (ABI).
Clinical details and investigations in five patients with MELAS
Results of sequencing five patients with MELAS
Results
Southern blotting excluded a large scale rearrangement of mtDNA in all patients. Systematic sequencing of all tRNA genes and the three mitochondrially encoded COX genes was undertaken. In patients 1, 2, 4, and 5 changes were identified compared with the Anderson sequence19 and these are listed in table 2. None of these changes were likely to be of pathogenic relevance, either because they had previously been shown to occur in a control population or because they were silent not producing amino acid substitutions.
In patient 3 we identified a G to A transition at position 583 in the aminoacyl acceptor stem region of the tRNA gene for phenylalanine (figs 1 and 2). This mutation was identified in heteroplasmic form in muscle from the biopsy aged 12 years but was not detected in mtDNA extracted from the patient’s blood sample taken at the age of 24 or from her mother’s blood (fig 1). The proportion of the G583A mutation in the patients muscle was 58%. Skin fibroblasts obtained form the patient did not harbour the mutation. The mother was well and had not had a muscle biopsy and the patient had no siblings. This mutation was not identified in mtDNA extracted from the blood of 100 healthy controls or from the muscle of a further 60 patients with ragged red fibres on muscle biopsy and varying mitochondrial disease phenotypes. Evolutionary comparison showed that a G nucleotide at position 583 is highly conserved.45
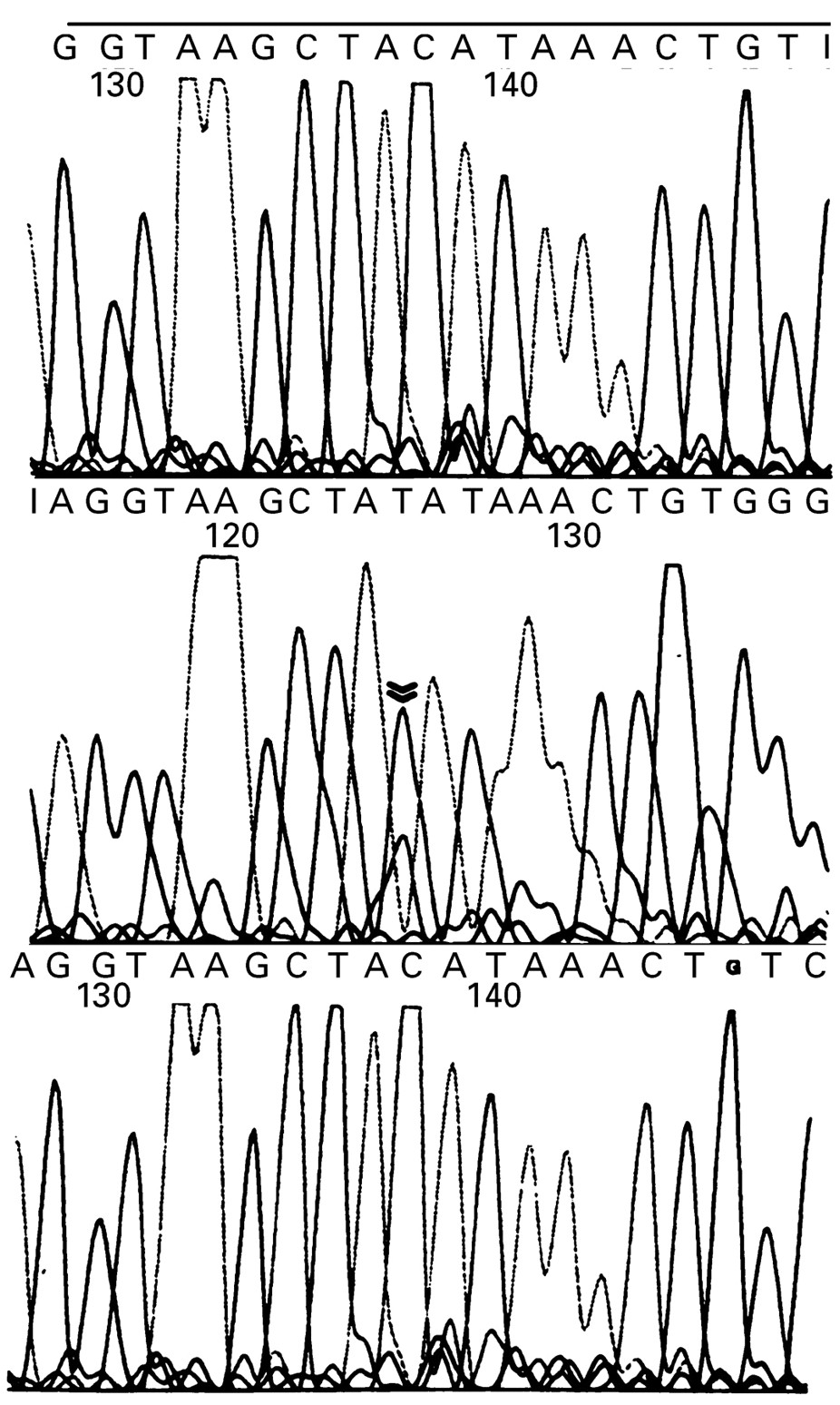
Chromatogram showing the heteroplasmic G583A (C to T on the antisense strand shown) transition in the mitochondrial tRNA gene for phenylalanine. Top panel; normal control blood, middle panel; patient 3 muscle, bottom panel; patient 3 blood. Double arrow indicates the mutant nucleotide, which was only detected in the patient’s muscle.

{kind=link}
{kind=link}
Theoretical two dimensional structure of the tRNA phenylalanine molecule illustrating the G583A transition mutation.
Discussion
The acronym MELAS was introduced by Pavlakis et al 2 to characterise a specific group of patients who had the onset of symptoms between the ages of 3 and 11 years. They met the following criteria: normal early development, short stature, seizures and alternating hemiparesis, hemianopia, or cortical blindness. They all had ragged red fibres, lactic acidaemia, and parieto-occipital lucencies on CT. It was suggested that MELAS was a distinct clinical entity separable from MERRF (myoclonus epilepsy with ragged red fibres) and the Kearns-Sayre syndrome (KSS). Hirano et al 20 proposed the following “invariant“ criteria for a diagnosis of MELAS; (1) stroke-like episodes before the age of 40 years, (2) encephalopathy characterised by seizures, dementia, or both, and (3) lactic acidosis, ragged red fibres, or both. They suggested that the diagnosis is secure if there are at least two of the following; normal early development, recurrent headache, or recurrent vomiting. They did note that the distinction between MELAS and MERRF or KSS was not absolute.2 Many more cases of MELAS have now been described and it is clear that it is the clinical and radiological features of the stroke-like episodes themselves which are the cardinal features of the MELAS phenotype.21 22The additional features represented in the acronym are variably present and there may be other features of respiratory chain disease which fall outside this strict definition. For example, lactic acidaemia and ragged red fibres, although often associated, are not always present.6 The age of onset is variable and cases in which the first stroke-like episode occurred long after the age of 40 have been reported.23
The variation in natural history and clinical features of patients with strokes typical of those described in MELAS is well illustrated by the cases reported here (table 1). Two of the patients—1 and 5—only had single events and were followed up for 7 and 8 years after the initial stroke respectively with no further events. Patients 4 and 5 illustrate that the stroke-like episodes can be many years apart. By contrast, patient 2 presented for the first time at the age of 52 years with a catastrophic illness leading to death in 6 months. In three patients (2, 4, and 5) there were other clinical clues to suggest a mitochondrial respiratory chain disorder before the stroke-like episodes, namely preceding deafness, cataracts, or optic atrophy. It is notable that patient 5 evolved radiological and clinical features of Leigh’s syndrome as well as MELAS. This combination has, to our knowledge, not been described before and highlights the overlap which can be seen between apparently distinct phenotypes of respiratory chain disease.24
From a practical viewpoint these findings indicate that the diagnosis of MELAS (or perhaps more accurately a mitochondrial stroke) should be considered at any age in the presence of consistent clinical and imaging features. Such features should prompt a search for other clinical and historical features suggestive of a respiratory chain disorder which may be varied, subtle, or completely absent. The most useful investigations are a muscle biopsy, with appropriate staining, and mtDNA analysis.25
Most patients who exhibit features of the MELAS phenotype harbour a point mutation at position 3243 in the tRNA gene for leucine (UUR).6 Five further point mutations in the same tRNA gene have been described in association with MELAS.26-30 This has led to the suggestion that defects in this gene are specifically associated with this phenotype28 possibly by affecting RNA processing.31 However, subsequent studies have shown that defects elsewhere in the mitochondrial genome can also be associated with MELAS. These include point mutations in the tRNA genes for valine and cysteine and in the COX III gene and large scale rearrangements (table 3).32-34
Pathogenic MtDNA mutations associated with stroke-like episodes
The five patients described here had at least one typical stroke-like episode, COX positive ragged red fibres and lactic acidaemia and did not harbour a known mtDNA mutation. None of these patients harboured SDH positive blood vessels in muscle as have been described by others.41 In view of the association between tRNA gene defects and the presence of ragged red fibres42 and the finding that a mutation in a COX gene can associate with MELAS,36 we systematically analysed all tRNA and mitochondrial COX genes.
There are three lines of evidence to suggest that the new mutation we identified in patient 3 is pathogenic. Firstly, it is heteroplsmic, a feature generally associated with disease causing mutations rather than neutral polymorphisms.17 Secondly, it occurs at a highly conserved position within the tRNA gene for phenylalanine, suggesting functional importance of the wild type nucleotide at this position. Thirdly, we did not identify this mutation in a large panel of normal controls or in a further 60 patients with mitochondrial phenotypes who do not harbour any known mutations. Further confirmation of its pathogenicity requires functional studies and the identification of this mutation in additional kindreds. Unfortunately we were unable to perform functional studies as the mutation was not present in fibroblast cultures from this patient and myoblasts were unavailable. One other mutation in the tRNA gene for phenylalanine has been described in a patient with a myopathy and acute rhabdomyolysis indicating that different phenotypes may be associated with mutations in this gene.43 The absence of the 583 mutation from the mother’s blood may be because the mutation arose de novo in the patient or because it has been lost from blood, possibly because of a selective disadvantage of blood cells harbouring the mutation.44 If the mother does harbour the mutation we presume it is in an insufficient amount to produce disease. It is notable that the patient did not have clinical evidence of a myopathy and only had mild histopathological changes in muscle. In view of her clinical presentation it is likely that she harbours higher amounts of this mutation in her brain.
The absence of mutations in the other four patients indicates that mitochondrial proliferation and formation of ragged red fibres are not necessarily associated with tRNA gene defects. It is likely that these patients harbour mutations elsewhere in the mitochondrial genome. The possibility that they harbour nuclear gene mutations cannot be excluded, although this seems less likely as ragged red fibres seem to associate with primary mitochondrial DNA defects.
In conclusion, we described the clinical and molecular genetic findings in five patients with stroke-like episodes typical of the MELAS phenotype. We provided evidence that a point mutation in the tRNA phenylalanine gene can associate with MELAS and that there is likely to be further genetic heterogeneity underlying the stroke-like episodes associated with mitochondrial respiratory chain disease. It is suggested that “mitochondrial stroke” may be a more clinically useful term.
Acknowledgments
We thank Professors Miller and Hodges and Drs Fish, Clarke, and Pilling for allowing us to study patients under their care. Support from the Medical Research Council of Great Britain (MGH) and the Muscular Dystrophy Group of Great Britain and Northern Ireland (IPN) is gratefully acknowledged.