Article Text

Abstract
BACKGROUND Striatal necrosis has been related to various clinical syndromes, with acute or chronic progression, and juvenile or late occurrence, but the most common type is Leigh’s encephalopathy.
METHODS Between 1967 and 1995, six out of seven related patients with chronic familial dystonia were examined. MRIs were performed in four, between 1992-1994. The seven members, affected over three generations, were the father, three daughters (one surviving), and three surviving grandsons.
RESULTS The leading symptoms were gait disorders and dystonia in all, dysarthria in six, verbal and motor stereotypies in two, and parkinsonian and cerebellar signs in three. Optic neuropathy was found in three. A frontal lobe syndrome without amnesia occurred in two. Symptoms occurred between the second and the fifth decade, with progressive deterioration. Magnetic resonance imaging, performed in four, showed in the two patients with severe neurological signs diffuse striatopallidal abnormal hyposignal (comparable with CSF signal) in T1 weighted images, suggesting extensive necrosis of the striatum and pallidum, associated with thalamo-subthalamo-rubro-dentato-nigral and substantia innominata hypersignals in T2 weighted images suggesting gliosis in these respective areas. The same images were described to a lesser extent in a third patient. Concentrations of lactate in CSF and serum were normal in three. Muscle biopsy, performed in four, was shown to be normal. Enzyme histochemistry showed complex I, III, and IV deficiency in surviving patients.
CONCLUSION This familial dystonia of chronic progression may be related to basal ganglia necrosis or gliosis, associated with alterations in the respiratory chain. These metabolic alterations probably play a part in the pathophysiology of these unusual brain lesions.
- striatal necrosis
- familial dystonia
- dysarthria
- mitochondrial disorders
Statistics from Altmetric.com
Familial dystonia is often essential but the occurrence of striatal lucencies suggests other entities including a large group of metabolic disorders, including mitochondrial defects.1 2Striatal necrosis was first described by Paterson and Carmichael, in young children.3 Later, some reports have described similar lesions in adults,4 5 associated with different clinical pictures.6 7 Striatal necrosis had appeared as a common feature of subacute encephalopathy of Leigh’s type.5 8 Its relation to mitochondrial disorder has been ascertained,2 but mitochondrial diseases which represent a chemically defined entity, correspond, in fact, to an association of very different clinical syndromes, including pure encephalopathy, neuropathy, myelitis, or myopathy.6 9 To our knowledge, familial late onset dystonia with diffuse lesions of the basal ganglia has not previously been reported in mitochondrial diseases. Analysis of the current literature disclosed only one family of late onset dystonia with dysarthria and striatal hypodensities, reported by Druschky10; however, he did not study the respiratory chain complexes, nor the muscle biopsy. We report seven family members with dystonia. Six of these have been examined or followed up by one of us between 1967 and 1995.
Case reports
Figure 1 gives the relation between each relative, and table 1summarises the main clinical features.

Family tree. ○=Female; □=male; /= deceased; patients are shown in black.

Case 7: nigral high intensity signals are bilateral, associated with a punctiform high intensity signal in the red nuclei.
Summary of clinical presentation
CASE 1: PROPOSITUS I.4
His daughter (case 4) and grandson (case 5) remember that he had an abnormal gait, with probable dystonia in lower limbs. In 1914, he became divorced after his wife gave birth to a daughter (case 2). He was the father of a second daughter (case 3), before he married again. Later, he had five other children. The different family names of his children explain the delay before recognising that they were relatives. He died at the age of 54, from sequelae of poison gas during the first world war, and was not necropsied. Before he died, he was able to walk only with difficulty due to dystonia and equinism.
CASE 2: PROPOSITUS II.2
This woman complained of gait disorders and dystonia at the age of 46. No somatic disorder occurred before this period, but she had psychiatric symptoms with personality disorders. She was admitted to hospital at the age of 56. At this time, she was unable to walk, because of pronounced dystonia and cerebellar ataxia. Speech was slurred. At rest, a mild hypertonia appeared in all limbs. Rigidity was considerably increased during voluntary movement and was then associated with torsional spasms involving both legs, with equinism. Alternating movements of the hands disclosed bradykinesia. No treatment had improved the dystonia nor the rest hypertonia. The predominant features were dystonia occurring during voluntary movements, parkinsonism, and dysarthria. She died at the age of 71. A necropsy was not performed.
CASE 3: PROPOSITUS II.3
This woman had moderate gait disorders. She has been examined once in 1968. At this time, she had gait unsteadiness, with falls. Speech was explosive. No dystonic posture occurred but athetoid movements were seen in both hands. Deep tendon reflexes were brisk. She did not complain of these disorders. The axial cerebellar syndrome and abnormal movements were associated with mental deterioration. She died at the age of 67. A necropsy was not performed.
CASE 4: PROPOSITUS II.8
She had been a nurse until the age of 58 but she had complained of gait disorders at the age of 51. She was first examined at the age of 63, in 1992. The most striking feature was that she waddled, despite the fact that there was only mild spasticity in the legs, and mild dystonic posture in the toes of the right foot. Strength was slightly reduced proximally in both legs. No parkinsonian symptoms were present. Speech was nasal and monotonous with normal volume. Neuropsychological studies included the Wechsler memory scale,11 and parts of the Wechsler adult intelligence scale (WAIS; cubes and similitudes),12 Buschke test,13 and digit span. They showed impairment for recall of a list of words, improved by indexation (Buschke test): free recall 1: 5/16, delayed free recall: 7/16. Nerve conduction velocities (NCV), somaesthesic evoked potentials (SEP), EMG, and EEG were normal. Brain CT and MRI of the spine were normal. In 1995, MRI of the brain with T2 weighted images (1Tesla, Magnetom impact, TE = 14, TR = 3200, slice thickness = 5 mm) showed diffuse abnormal hypersignal of both putamen, associated with abnormal hypersignal involving both substantia nigra. Pallidum and red nucleus density seemed to be normal. Muscle biopsy was performed when she was 62 and showed isolated ragged red fibres, corresponding to 3% of total fibres. Electron microscopy showed normal mitochondria, without inclusions.
CASE 5: PROPOSITUS III.10
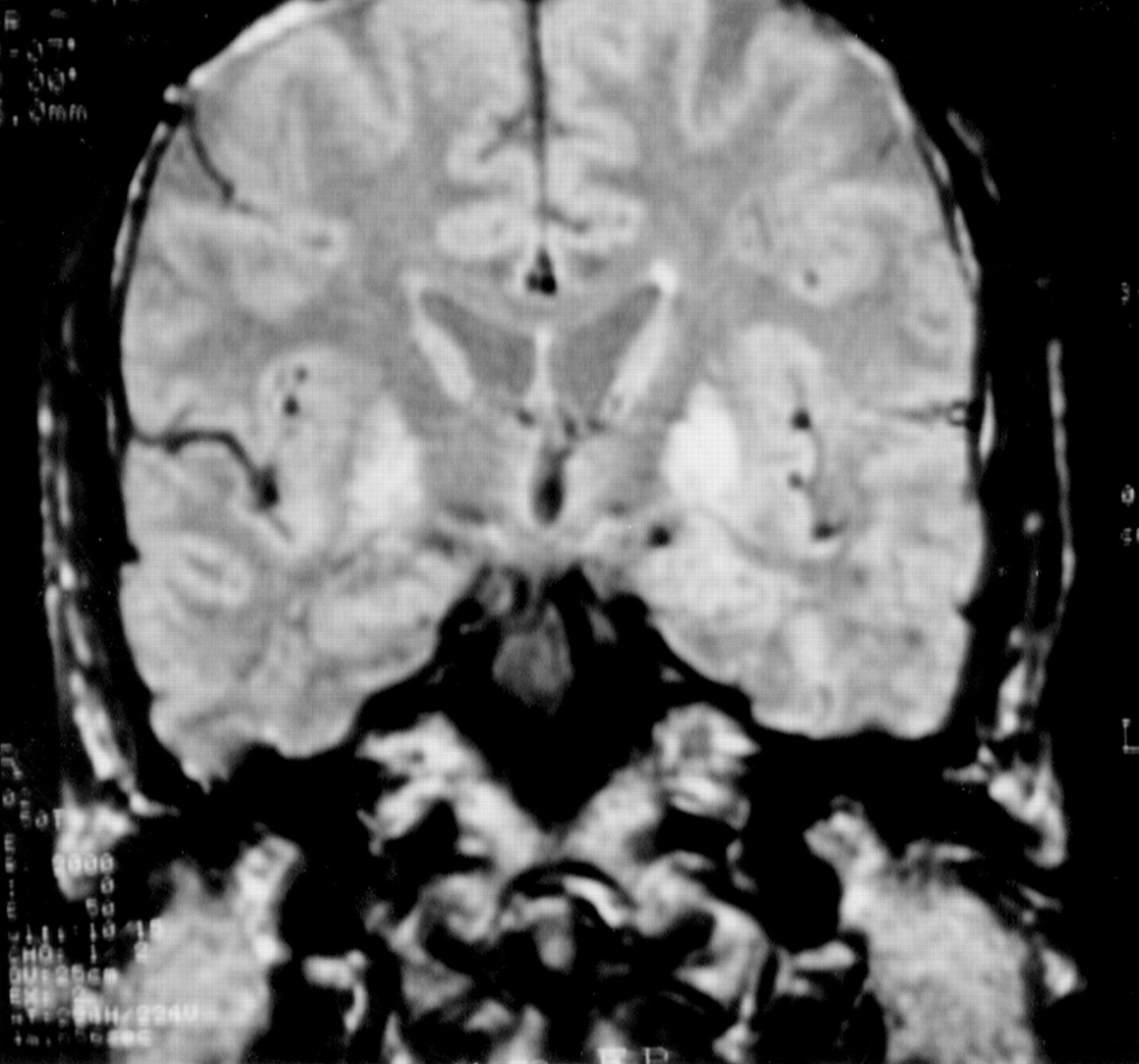
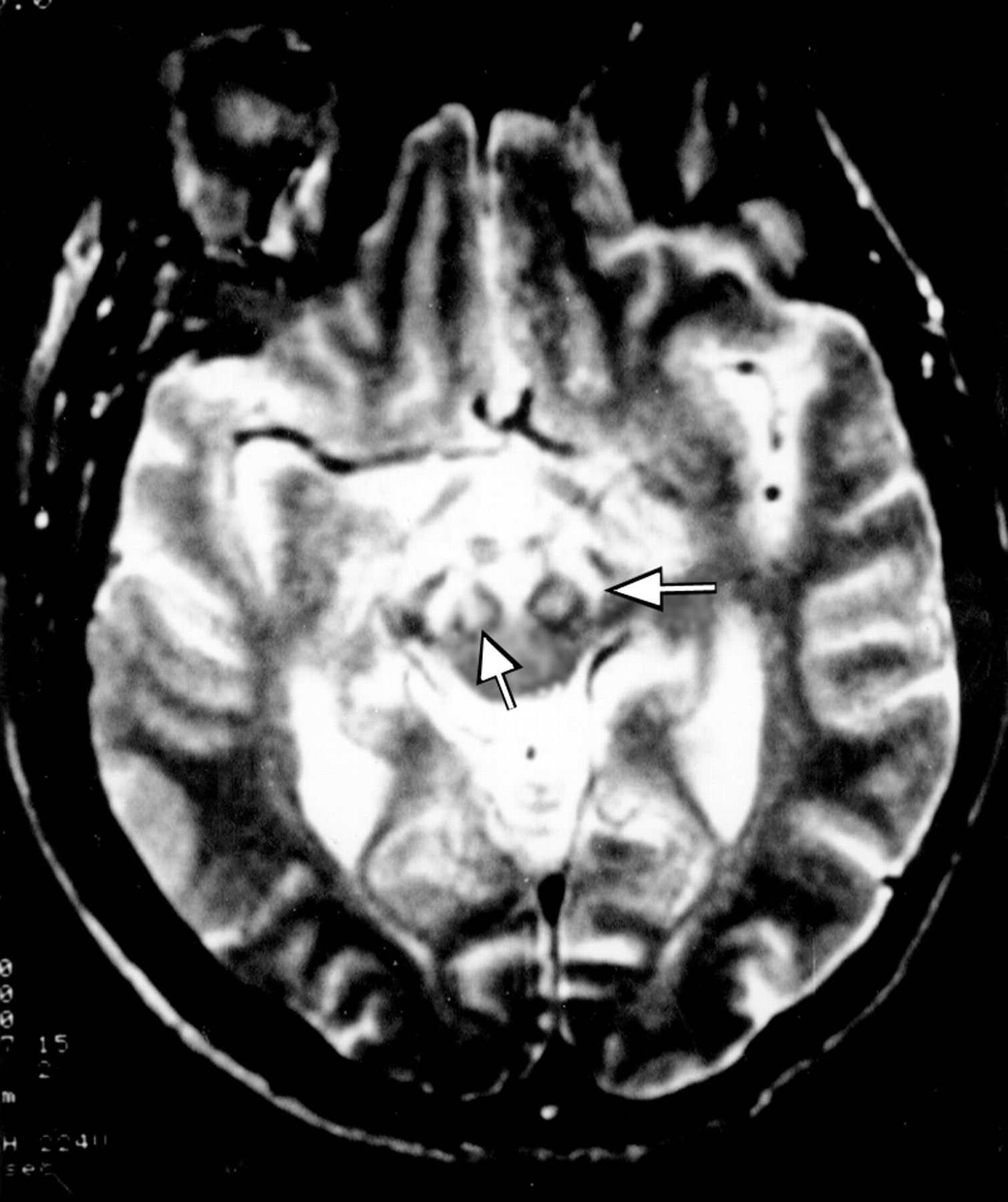
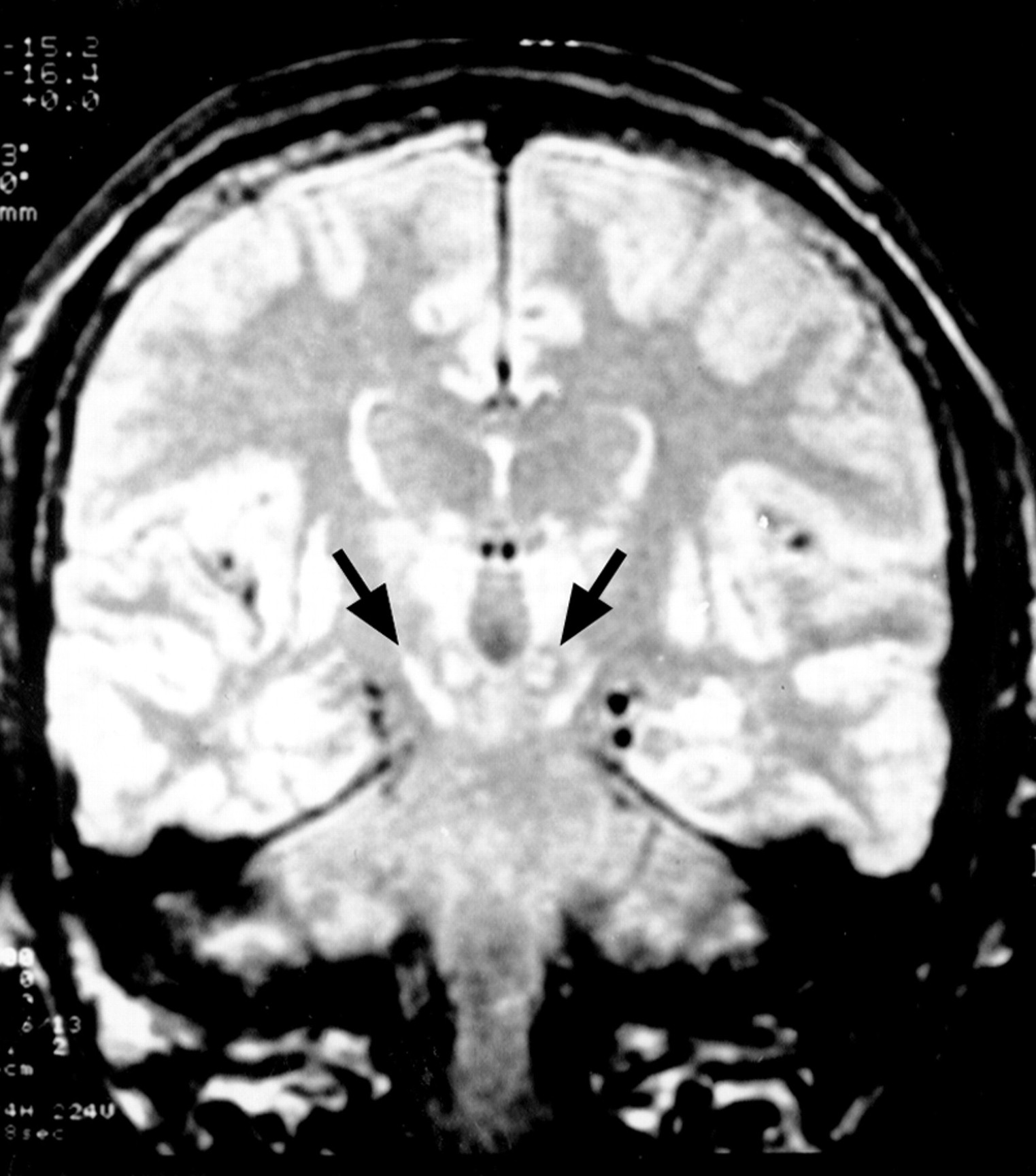
This first son of case 3, born in 1935, had been a draughtsman until the age of 33. Birth and childhood had been normal. At the age of 27 years, he noted the appearance of mild gait disorders which progressively worsened. He was admitted once in 1967, for impaired balance, dyskinesia, dysarthria with hypophonia, and swallowing impairment. At rest, hypotonia was permanent. During voluntary movements, hypertonia due to dystonia with myoclonus occurred. Dystonic movements interfered with the voluntary, precise, and rapid alternating movements of the hands, performed for bread buttering, hairdressing, drumming, or scratching. Walking was stiff, unstable, and also impaired after a few minutes by dystonic posture of the left foot. He was amimic, with monotonous speech and micrographia. There were no pyramidal signs, nor oculomotor deficit. One year later, falls occurred daily. Dystonic dysarthria worsened until speech was no longer understood in 1973. Aphonia extended to severe hypophonia in 1976. Blepharospasm occurred in 1978, and worsened progressively. It has been partly improved by repeated injections of botulin toxin, which have been continued. Other treatments orally administered (dopaminergic agonists, anticholinergic drugs) were not effective on motor disorders and were stopped. In 1995, the patient was still able to walk, with moderate dystonic posture of both feet, unstable gait, axial akinesia, taking shuffling steps, without arm swinging. He had moderate hearing loss. Neuropsychological evaluation was limited by aphonia. Verbal testing was performed using a small computer. Premorbid intelligence quotient (IQ) determined by Beauregard’s automatisms14was 36/40, which corresponds to high IQ (130). Verbal memory testing from the Signoret memory battery15 was at the lower limit of normality (6/12, controls: 8.31 (SD 1.25)). Digit spans were normal16 (direct: 7, inverse: 5). Progressive matrices of Raven (PM 47)17 were normal (A: 11, B: 12, C: 9, time: 9 min 48 s). EEG, EMG, and NCV were normal, as was the muscle biopsy. Ophthalmological examination showed normal visual acuity and field, and a normal electroretinogram. Visual evoked potentials (VEPs) were bilaterally disorganised with decreased velocity, suggesting a mild optic neuropathy. T1 weighted coronal and axial images MRI on (TE = 12, TR = 400) showed diffuse putaminopallidal abnormal hyposignal, comparable with CSF signal (fig 2), and to necrotic cavities, suggesting extensive necrosis of the striatum and pallidum. In T2 weighted axial images (TE = 100, TR = 2000), the putaminopallidal area appeared as a large hypersignal (similar to the CSF signal). This intense abnormal signal involved the whole putamen and pallidum, and the caudate nucleus was involved to a lesser extent. A mild high intensity signal was also seen in both thalami (fig 3). At the level of the brain stem, T2 weighted axial images showed symmetric abnormal hypersignals involving the whole substantia nigra. An abnormal hypersignal was also located in both red nuclei, surrounded by a lower signal, bilaterally (fig 4). The coronal plane confirmed red nuclei lesions, and suggested symmetric implication of subthalamic nuclei and below of both substantia nigra (fig 5). This also showed an abnormal hypersignal surrounded by hyposignal in the cerebellum, bilaterally, in the area of dentate nuclei (fig 6). An abnormal signal was located below the pallidum and above the optic tracts in the area of the substantia innominata, probably including the Meynert’s basalis nuclei, according to the atlas of Nieuwenhuys et al 18 (fig 7). This may suggest a gliosis in these respective areas : substantia nigra, thalami, red nuclei, dentate, and basalis nuclei. SPECT, using99mTc-hexamethyl-propylene-amine-oxime (Hm-PaO) showed severe symmetric striatal hypoperfusion with bilateral prefrontal hypoperfusion.

Case 5: MRI with T1 weighted coronal images shows diffuse abnormal putaminopallidal low intensity signal, comparable with the CSF signal.

Case 5: in T2 weighted axial images, the putaminopallidal area appears as a large high intensity signal. The caudate nucleus is involved to a lesser extent. A mild high intensity signal also appears in both thalami.

Case 5: T2 weighted axial images show symmetric abnormal high intensity signals involving both substantia nigra (left arrow), and both red nuclei (right arrow), which are surrounded by a lower intensity signal.

Case 5: the coronal plane confirms the involvement of red nuclei, medially to the subthalamic nuclei and above both substantia nigra, which were also damaged.

Case 5: posterior coronal plane shows an abnormal signal surrounded by low intensity signal in the cerebellum, bilaterally, in the area of the dentate nuclei (arrow).

Case 5: an abnormal signal is located below the pallidum and above the optic tracts in the area of the substantia innominata (arrow).
CASE 6: PROPOSITUS III5
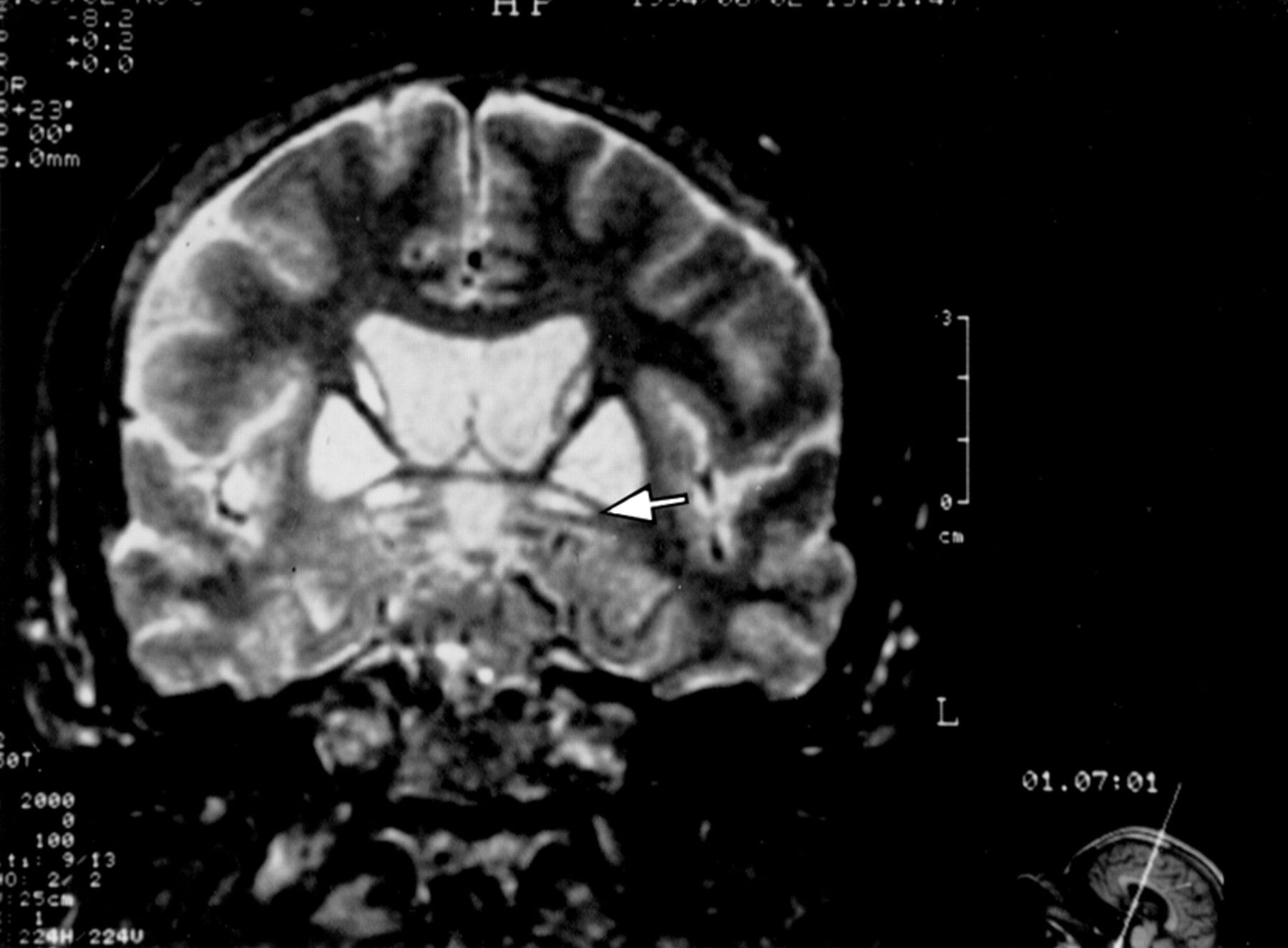
This third child of case 2, born in 1944, had been an engineer until the age of 32. He complained of gait disorders at the age of 24 years. In 1970, at the age of 26, neurological examination showed dystonia. At rest, there was no abnormal posture. Walking induced progressive distorted posture of the feet, with equinism, whereas hypotonia was seen at rest. During movement or posture of the arms, athetoid movements involved the left hand. Handwriting cramps were also noted. These disorders worsened very slowly during the next five years, with gait impairment and progressive development of cognitive disorders with a frontal lobe syndrome. In the 1980s, stereotypies developed and progressively worsened. The patient was admitted in 1994, after dystonia worsened. Dystonic postures prevented the patient from walking further than 10 m. The frontal lobe syndrome worsened with permanent palilallia, and verbal and motor automatisms involving mainly the upper limbs. Speech was dystonic with stuttering. Writing was dystonic and macrographic. Longlasting treatment using anticholinergic drugs and dopaminergic agonists did not modify the motor status. The mini mental state examination result19(MMSE) was 28/30. Neuropsychological study showed mild impairment in verbal and visual memory (low recall of a list of words from the Signoret memory battery:15 6/12, impairment of Rey figure recall:20 10/36), whereas visuoconstructional abilities were normal (Rey figure copy). Premorbid IQ14 was 125. Fluency21 22 was low (category: 10, letter (C): 5). The results of the Wisconsin card sorting test23 (WCST) were abnormal: only two categories were defined, with 32 errors (including 24 perseverative errors). EEG, EMG, and NCV were normal, as was the muscle biopsy. Visual evoked potentials were altered, with increased latency. Brain CT disclosed bilateral hypodensities involving the whole putamen and pallidum. Density analysis, using a workstation (IBM compatible computer, PIP, and SM matrox cards, original software of density analysis) showed that both striatopallidal hypodensities had the same density as the CSF in the ventricles (89 density units (DU)/ 255 DU). T1 MRI showed diffuse striatopallidal abnormal hyposignal (fig8). In T2 images, abnormal hypersignals were seen in both substantia nigra and red nuclei, as in patient 5. Despite sedation, the patient had moved during the MRI study. The poor quality of images did not allow assessment of other abnormalities. SPECT with HmPaO showed diffuse prefrontal and lenticular hypoperfusion.

Case 6: T1 images show diffuse abnormal striatopallidal low intensity signal.
CASE 7: PROPOSITUS III.7
This sixth child of case 2, born in 1949, was a highschool teacher, until the age of 45. In 1993, he complained of writing impairment associated with abnormal movements of the right hand, dysarthria, and dysphagia. Neurological examination disclosed permanent athetoid movements of the right hand and fingers. There was right hypotonia at rest, and dystonia with hypertonia during movements. Writing was micrographic. There was left adiadocokinesia. Speech was slurred and dystonic. Constant grimacing occurred because of permanent dystonic movements of the face. Walking was slightly impaired by dystonic posture in the right foot. Symptoms worsened progressively during the next year, with the development of permanent athetoid movements in the right hand. The patient was administered levodopa with decarboxylase inhibitor (DCI), and dopaminergic agonists but no change occurred. Neuropsychological study showed normal verbal and visual memory. Fluency was low (letter: 9; category: 14). The Mattis results were low: 122/144. The patient failed to perform motor sequences, and the ability to define concepts was reduced. The trail making test was impaired in time (A: 56 s, B: 185 s), whereas the results of the Wisconsin card sorting test (WCST) were normal. EEG, EMG, and NCV were normal, as was the muscle biopsy. Visual evoked potentials were altered. Brain CT showed small bilateral hypodensities in both the putamen and pallidum, prevailing in the left. T2 weighted MRI of the brain showed abnormal hypersignal in the striatum, and in the right substantia nigra. After one year, hyperintensities had increased in both pallidoputaminal areas (fig 9). Bilateral abnormal hyperintensities also involved the substantia nigra and the red nuclei in which a punctiform hypersignal was seen (fig 9). SPECT showed prefrontal hypoperfusion, prevailing in the medial regions, bilaterally.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Case 7: one year after clinical onset, the lenticular hyperintense signals are increased in size, involving the pallidum and the putamen, although the dorsal portion of the right putamen appears less involved.
GENERAL STATUS
Of the six patients examined, none had somatic evidence of mitochondrial disorders: there was no cardiac or respiratory failure, no auditory or muscular involvement, and no ophthalmoplegia, and no hepatic or renal failure. None of the patients had a history of toxic misuse, or poisoning by methanol, thiocyanate, or 3-nitropropionic acid. Clinical and biological studies failed to show symptoms suggesting Wilson’s disease.
In all alive patients, CSF and blood studies were normal. Lactate concentration was normal in serum and CSF at rest, but increased abnormally in two patients (6 and 7) during and after physical exercise. In the other patients, physical exercise could not be performed. The methodology of muscle biopsy and respiratory chain compound study has been described previously.24 The assays were performed on frozen muscle samples kept in dry ice or at −80°C, obtained by a surgical technique from deltoid muscle. Parts of the muscle biopsies were serially sectioned and then subjected to a histochemical staining battery including the modified Gomori trichrome technique, Cytochrome oxydase (COX), nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR), succinate dehydrogenase (SDH), and myosin-adenosine triphosphate (ATPase). The percentage of ragged red fibres or COX negative fibres recorded for each patient was below 3% except in the oldest patient. Electron microscopy was normal in all studied cases. Respiratory chain compounds were detected by spectrophotometry. The analysis included NADH ubiquinone oxidoreductase (complex I) measurement with and without rotenone in parallel cuvettes, succinate cytochrome c oxidoreductase (complexes II + III), ubiquinol cytochrome c oxidoreductase (complex III) measured with and without antimycin A in parallel cuvettes, cytochrome c oxidase (complex IV), and citrate synthetase (DTNB as acceptor). With the parallel cuvettes, the technical conditions of analysis are identical. The results were compared with those of 29 normal controls (table 2). The total DNA was extracted from small parts of frozen muscle, digested by a restriction enzyme, run on agarose gel, and transferred to nitrocellulose. Mitochondrial DNA of muscle was analysed by Southern blot using two probes at the same time: total mitochondrial DNA extracted from human placenta and CDNA probe of the human 28 S gene. In all cases, total complex I was reduced. After the rotenone step, the complex I was below the lower value of controls in all cases, significantly reduced in cases 4 and 6. The complexes II, III, and citrate synthetase results were not available in patient III.10 because of impaired conservation. Complex I and complex III were decreased in the other three patients, in comparison with controls. Complex IV was decreased in two. Complexes II + III were normal. Mitochondrial DNA study was normal in all cases, failing to show any deletion or known mutation.
Studies of respiratory chain compounds and mitochondrial DNA
Discussion
We have isolated a new clinical syndrome of familial dystonia. Clinical and radiological features suggest a new entity which differs from previous descriptions of mitochondrial diseases.25-28 Despite an apparent clinical heterogeneity, all of our patients were affected by dystonia and dysarthria. The dysarthria was assessed in six patients. The prominent feature was the dystonia occurring only during movements, and disappearing completely at rest. The same biochemical disorder, and probably the same brain lesions, had induced strikingly different clinical patterns, with various degrees of cerebellar and parkinsonian syndromes. Of the two most disabled patients displaying the same MR images, parkinsonian signs were predominant in one, while dystonic and cerebellar signs were predominant in the other. Athetoid movements occurred in cases 3 and 7, who were not directly related, whereas dystonia was prominent in the others. Frontal lobe syndrome occurred in three patients, whereas none of these patients had all of the specific criteria for the diagnosis of dementia. A severe behavioural impairment of the frontal lobe type was seen in only one of the surviving patients (case 6). The most impressive feature was his permanent motor and verbal automatisms. In case 7, the brother of case 6, the frontal lobe syndrome involved only instrumental skills. The most affected test was the motor sequences, whereas other tests (including WCST) were normal. Graphic sequences were normal. The specific impairment of motor sequences may reflect a subtle frontal lobe syndrome. Nevertheless, these dissociated results could also be explained by a different sensitivity of frontal lobe tasks. The syndromic association of action dystonia, with frontal lobe symptoms or athetoid movements, without dementia is uncommon in the current literature. We have shown evidence that this syndrome is related to a mitochondrial cytopathy because all of our patients had a complex I defect which appeared either alone, or associated with complex III or IV defect. These partial defects of complexes I, III and IV were associated with a unique pattern of deep brain lesions involving only the basal ganglia, which may be compared only to Leigh’s disease and related rare mitochondrial disorders.
The long-lasting course of the disease could have suggested degenerative process such as luysopallidonigral atrophy, which is a familial inherited disease, disclosed by rigidity, parkinsonism, and cognitive impairment.29 Other related syndromes, with involvement of the pyramidal tract, thalami, or dentate nuclei, have been described,30-32 but none of them displayed putaminal lesions, nor spongiosis (or necrosis) of the lenticular formation. The clinical syndrome that we have described is different from previously described mitochondrial encephalopathy which usually induces multivisceral failure. Common phenotypical syndromes involving the CNS: Leber’s disease, ophthalmoplegia plus syndromes, and Leigh’s disease, could be compared with our cases, as they displayed either parkinsonian syndrome or striatal lesions. However, many extrapyramidal syndromes associated with mitochondrial disorders have not been clearly classified, because of their polymorphism.6 The long-lasting predominance of movement disorders is unusual in mitochondrial diseases.6 27 Different symptoms of mitochondrial encephalomyopathy were missing in our patients. There was no peripheral neuropathy, either clinically or electrophysiologically. There were no seizures, nor ophthalmoplegia. Previous anatomical, CT, or MRI case reports of lenticular lesions have been reported and may be compared with our cases. Leigh’s disease, a heterogenous entity defined by subacute necrotising encephalomyelopathy occurring in children, has also been reported in adults, with chronic evolution.4 5 It is usually sporadic, but autosomal dominant forms have been reported.33 This condition differs from our patients as the pallidum is inconsistently involved, the red nuclei is spared, and the periaqueductal grey matter is usually destroyed.5 In Leber’s disease, which is transmitted by females, optic atrophy is prominent, but dystonia has been reported by many authors, associated with putaminal lucencies.34-36However, an overlap between these diseases is probable.37Anatomical study in Leigh’s disease, reported by Garcin et al, obviously showed involvement of Meynert’s basalis nucleus, as in our patients.38 The family described by Druschky10 had the same clinical features as ours: long-lasting dystonia, with dysarthria and autosomal dominant transmission, but both MRI and anatomical study confirmed that the pallidum was not involved. In our two most disabled patients, MRI showed lenticular and caudate hyposignals, suggesting necrotic cavities or spongiosis. In case 7, MRI follow up showed that the pathological process started probably in the pallidum, the ventral part of the putamen and the substantia nigra, and was asymmetric. The muscle biopsies seemed to be normal morphologically but disclosed biochemistry abnormalities, and were then useful. The mode of inheritance seems to be autosomal dominant, whereas most mitochondrial diseases associated with mitochondrial DNA mutation or deletion are usually sporadic ot transmitted by women. In our family, the transmission suggested nuclear, rather than mitochondrial DNA alterations.
The pathogenic role of these metabolic defects in the development of the brain lesions remains to be proved. The implication of mitochondrial dysfunction in the pathogenesis of movement disorders has been demonstrated in several ways: (1) mitochondrial DNA mutations have been found in both unusual syndromes associating chorea and dementia,39 and subacute or chronic forms of striatal necrosis;40 (2) the dosage of respiratory chain complexes shows a specific decrease in complex I in the substantia nigra of parkinsonian patients,41 42 in multiple system atrophy, and sporadic or familial cases of late onset cerebellar degeneration.6 42 This suggests that the basal ganglia system is extremely sensitive to complex I defect. Different pathogenic factors have been discussed for the explanation of tissue lesions in mitochondrial disorders: infarction in anoxic tissue with secondary microangiopathy or primary involvement of small arteries. It has also been shown that mitochondrial toxins produce striatal excitotoxic lesions, by a mechanism involving energy depletion in vivo.43 The delayed occurrence of symptoms with chronic progression in our patients could be due to both partial defects in the mitochondrial respiratory chain, and the effect of aging. We did not show mitochondrial DNA mutation or deletion, which is not surprising in an autosomal dominant transmission. Different mutations have been found in Leigh’s syndrome, and other primary dystonias.40However, degenerative dystonia never displayed necrotic lesions involving the lenticular formation. Mitochondrial defects might be linked to toxins such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, and medications such as neuroleptic drugs.44 These causal factors could not explain the occurrence of the striatal necrosis of our patients, nor the clinical features, particularly the cerebellar signs.
In conclusion, we think that we have recognised a new clinical entity of hereditary mitochondrial striatopallidonigral disease which is expressed as a late onset progressive dystonia and finally involves the entire basal ganglia. The genetic defect underlying this disease still remains to be determined.
Acknowledgments
We are grateful to Dr A Lombes for the biological and mitochondrial study. We thank Dr AL Benabid for valuable comments and suggestions. We also thank Drs JP Pruvo and MM Ruchoux for their contribution to the radiological and pathological studies, and Dr E Boogusch for proof reading the manuscript.