Article Text

Abstract
Background: Mutations in the PTEN-induced kinase 1 (PINK1) gene have been identified in recessively inherited and sporadic early-onset parkinsonism (EOP).
Methods: A total of 131 Norwegian patients diagnosed with Parkinson’s disease were included. Of them, 89 participants had EOP (onset ⩽50 years); the remaining had familial late-onset disease (mean age at onset 64 years). PINK1 analysis included sequencing and gene dose assessment. Mutations were examined in 350 controls.
Results: Heterozygous missense mutations in PINK1 were found in 3 of 131 patients; none of the patients carried homozygous or compound heterozygous mutations. One of these three patients had a father affected by Parkinson’s disease, and he carried the mutation. Three new and seven known polymorphic variants were identified, although none seemed to be associated with disease risk.
Conclusions:PINK1 mutations are rare in Norwegian patients with EOP and familial Parkinson’s disease. However, the data suggest that some heterozygous mutations might increase the risk of developing Parkinson’s disease.
- EOP, early-onset parkinsonism
- PINK1, PTEN-induced kinase 1
Statistics from Altmetric.com
The causes of Parkinson’s disease are still largely unknown. Evidence suggests that both environmental factors and genetic susceptibility contribute to disease aetiology.1 Familial parkinsonism can be inherited as an autosomal dominant or recessive trait. Mutations in three genes have been associated with recessively inherited early-onset parkinsonism (EOP): parkin, DJ-1 and PTEN-induced kinase 1 (PINK1). Mutations in the parkin gene may account for nearly 50% of familial and a considerable proportion of apparently sporadic EOP (with age of onset ⩽45 years).2 Pathogenic mutations in the DJ-1 gene seem to be rare, causing <1% of EOP cases.3
Missense mutations in the PINK1 gene were first identified in three consanguineous Italian and Spanish kindreds affected with EOP.4 Mutations in this gene have now been found in families originating from several European and Asian populations, making PINK1 the second most common genetic known cause of EOP.5–9PINK1 mutations have also been identified in patients with sporadic disease, including heterozygous mutations of unknown pathogenicity.10,11 To further evaluate the pathogenic role of PINK1 mutations in familial and sporadic Parkinson’s disease, we performed a comprehensive mutation analysis of this gene in a series of Norwegian patients with Parkinson’s disease.
METHODS
Subjects
In all, 89 patients with EOP (age at onset ⩽50 years) were recruited from a study of the genetics of Parkinson’s disease in Central Norway. Mean (standard deviation (SD)) age at disease onset in this group was 44 (5) years (range 31–50 years); 27 of them had a family history of Parkinson’s disease. We also included 42 patients with familial late-onset Parkinson’s disease (mean (SD) age at onset 64 (6) years, range 51–75 years). Of them, 20 patients had a family history consistent with recessive inheritance, as broadly defined by the presence of parkinsonism in siblings or first-degree cousins, without evidence of affected parents or offspring. The remaining 22 patients had familial disease without a clear pattern of Mendelian inheritance. Of the 131 patients, 77 were men and 54 were women. In all, 350 healthy Norwegian controls were drawn from the same population (mean (SD) age 66 (13) years, range 49–100 years).
Patients were diagnosed with Parkinson’s disease in accordance with the Gelb criteria.12 Known carriers of homozygous or compound heterozygous mutations in the parkin gene were not included. All patients were previously screened for the presence of seven mutations in the LRRK2 gene, and mutation carriers were not included.13 Mutations in the DJ-1 gene have not been completely examined; several Norwegian patients with EOP were included in a previous study and no DJ-1 mutations were found.14 Informed consent was obtained from all participants, and the study was approved by the regional committee for medical research ethics in Central Norway and the Mayo Clinic Institutional Review Board.
Mutational analysis
All eight PINK1 exons were amplified by polymerase chain reaction, with primers flanking intronic sequences (primer sequences and assay conditions are available on request). Sequencing was performed using BigDye Terminator V.3.1. Sequence variations identified in patients were genotyped in 350 Norwegian control samples using designed TaqMan single-nucleotide polymorphism genotyping assays (Applied Biosystems, Foster City, CA, USA) or by direct sequencing.
Absolute quantitative polymerase chain reactions of PINK1 were performed using the iQ SYBR Green Supermix kit (BioRad, Hercules, CA, USA). Absolute quantification of template was obtained from a standard curve using the MJ Opticom Monitor V.3.1. For this assay the concentrations of PINK1 exon 4 and 7 were individually analysed and compared with concentrations of the external control gene, human serum albumin.
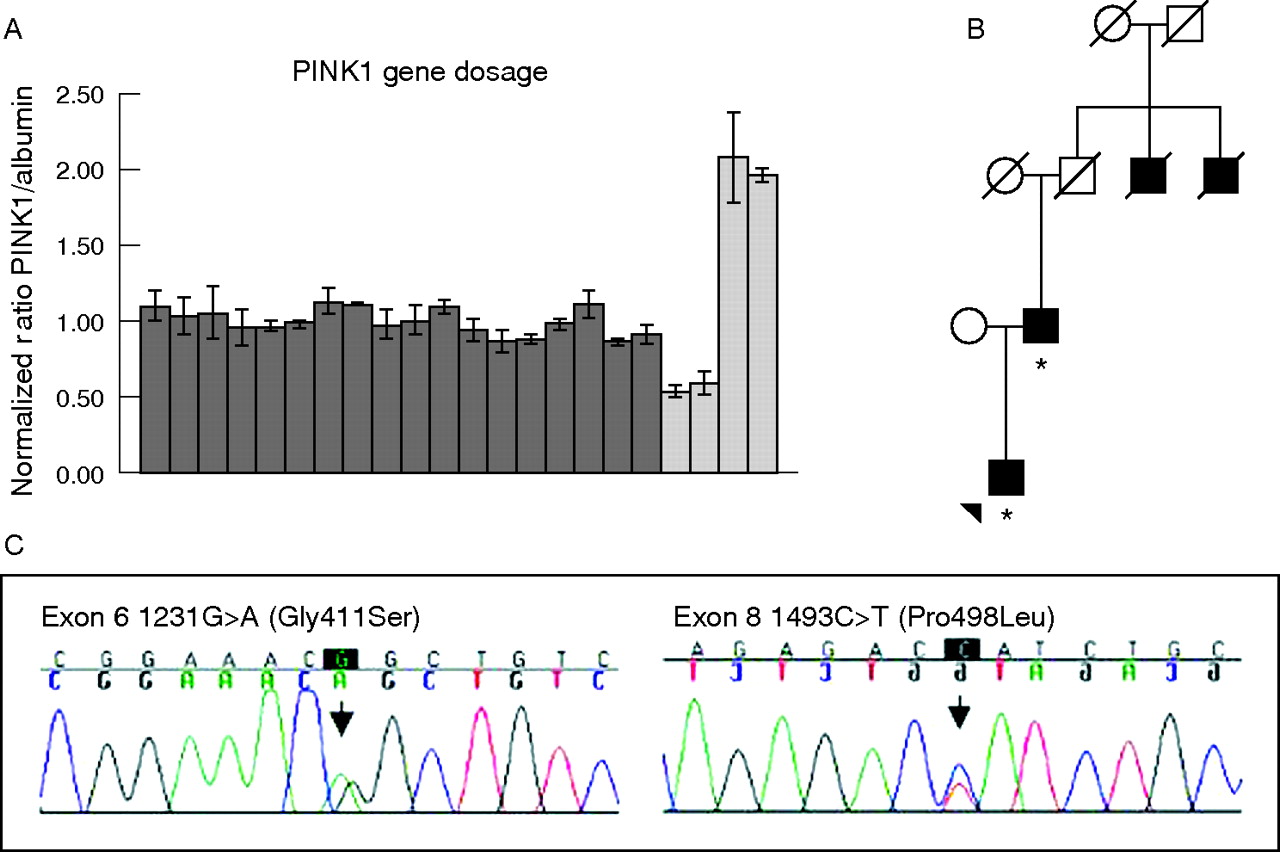
Each sample was run in a triplicate reaction. Relative gene dosage ratios with SDs were calculated by dividing the normalised mean PINK1 quantity by the mean albumin quantity. Positive controls for deletion and multiplication mutations were designed by using other amounts of DNA (fig 1A). A relative ratio with SDs between 0.75 and 1.25 was considered normal, a heterozygous deletion was expected at a ratio between 0.25 and 0.75, and a duplication was expected between 1.25 and 1.75. Ambiguous samples were re-run in triplicate with DNA from a separate tube.

{kind=link}
Results of PINK1 mutation analyses in Norwegian patients with Parkinson’s disease. (A) Relative ratios of gene dosage of PINK1 compared with albumin for 17 patients with early-onset parkinsonism (dark grey); the bars represent standard deviations. The ratios for two deletion controls and two triplication controls are shown in light grey. (B) Pedigree for the family of patient P392. The patient and his father are affected and have been tested for mutations in the PINK1 gene (denoted by *). Two paternal uncles of P392 were affected by Parkinson’s disease, but were deceased and thus not available for genetic testing. (C) Chromatograms showing the two potentially pathogenic mutations identified in this study.
RESULTS
Molecular findings
We identified two missense mutations in the PINK1 gene: c.1231G→A (Gly411Ser) and a novel c.1493C→T (Pro498Leu) mutation (fig 1C). Of the 131 patients, two were heterozygous for the Gly411Ser substitution and one carried Pro498Leu. These mutations were not found in any of the 350 controls.
We also identified three new and seven previously published polymorphic variants in PINK1 (table 1). None of the common polymorphisms were associated with Parkinson’s disease (p>0.05). Distributions of genotypes were in Hardy–Weinberg equilibrium. We identified one as yet undescribed exonic c.1745G→T mutation removing the stop codon (Stop582Leu), leading to the translation of nine additional amino acids until the next stop codon occurred. This variant was found in one patient and two controls and has unknown pathogenic significance. Two novel exonic variants were silent mutations. Both were found only in one patient each and were not present in control chromosomes. Deletions or multiplications of the PINK1 gene were not identified in any of the patients.
Polymorphisms and mutations in the PTEN-induced kinase 1 gene
Case reports
Patient P392 is a 45-year-old man carrying a heterozygous Gly411Ser mutation. His parkinsonian syndrome started at age 43 years, with bradykinesia, mild rigidity, resting tremor and a marked hypomimia. The bradykinesia on the left side is now severe. Dopaminergic treatment has so far not been started. The patient’s father has also recently been diagnosed with Parkinson’s disease at the age of 63 years, and sequencing of PINK1 exon 6 confirmed that he is also carrying the same mutation. Two of the father’s uncles had received a diagnosis of Parkinson’s disease (fig 1B).
Patient P371 is a 60-year-old woman who is heterozygous for the same Gly411Ser mutation. Age at onset in this patient was 50 years. She presented with typical asymmetric parkinsonism, with an excellent response to dopaminergic treatment. However, she developed severe levodopa-induced fluctuations, and subthalamic deep brain stimulators were introduced 1 year ago with a very good effect. There is no history of neurodegenerative disorders in her family.
Finally, patient P266 was diagnosed with Parkinson’s disease 4 years ago at the age of 58 years. He has a slowly progressive parkinsonian syndrome of mild bradykinesia, action tremor and rigidity. He carries a Pro498Leu mutation and has not yet been introduced to dopaminergic treatment. A paternal uncle was diagnosed with Parkinson’s disease, but he was not available for examination. The patient has a brother with an atypical action tremor without other signs of parkinsonism.
DISCUSSION
In this study, we performed a comprehensive mutation analysis of the PINK1 gene in a series of 131 patients. Mutations in the PINK1 gene seem to be rare in Norwegian patients diagnosed with Parkinson’s disease. We identified three heterozygous mutation carriers; none of the patients had homozygous or compound heterozygous PINK1 mutations. Deletions and multiplications of the PINK1 gene are uncommon, as only one exonic deletion has been reported.8 Also, our quantitative analysis failed to detect any such mutations.
Two patients were heterozygous carriers of a PINK1 Gly411Ser substitution. One of them (P392) has a family history of Parkinson’s disease, and his affected father carries the mutation, indicating a possible pathogenic role of this variant. Recently, a male patient with EOP starting at the age of 13 years was also found to be a heterozygous carrier of this mutation.15 A second previously unknown mutation, Pro489Leu, was found in a patient with late-onset Parkinson’s disease with a familial history of parkinsonism, but DNA from family members was not available for segregation analysis.
The clinical presentation of our three patients with PINK1-associated EOP was indistinguishable from idiopathic Parkinson’s disease and similar to that found in most other families and sporadic cases with PINK1 mutations.5,11 All patients had a slowly progressive parkinsonian syndrome; none of them showed signs of early dementia, had psychiatric symptoms or had dystonia at disease onset. Only one of our patients has received dopaminergic treatment and showed an excellent effect of levodopa treatment. She developed severe dyskinesias, which have been successfully treated with implantation of bilateral stimulators in the subthalamic nuclei.
The pathogenic significance of a single heterozygous PINK1 mutation is unclear. In most autosomal recessive disorders heterozygous carriers are clinically unaffected. However, there is evidence that heterozygous PINK1 mutation carriers might be at increased risk of developing parkinsonism. The two mutations found in this study were absent in a large number of Norwegian control chromosomes, making it less likely that they are rare polymorphisms. Both mutations are located in the PINK1 kinase domain and replace evolutionary conserved amino acids, and may thus affect the kinase activity.
A positron emission tomography study of unaffected heterozygous PINK1 mutation carriers indicated a reduction in F-dopa uptake in comparison with controls.16 Hence, PINK1 dysfunction might reduce striatal dopamine storage capacity and increase susceptibility to parkinsonism. Similarly, positron emission studies of mutation carriers in the parkin gene have shown a decreased F-dopa uptake compared with controls.17 Several studies have reported high frequencies of single-allele mutations in the parkin gene of patients with EOP, suggesting that single parkin mutations might be a risk factor for Parkinson’s disease.18 Interestingly, PINK1 has recently been shown to genetically interact with parkin in model systems, indicating that the two proteins act in a common pathway.19
PINK1 mutation screening of sporadic EOP cohorts from other European populations have identified findings similar to those in our Norwegian sample, with heterozygous mutations in patients that are absent in controls.10,11,20 In addition, six carriers of PINK1 mutations in a large German family with Parkinson’s disease presented with slight or mild symptoms of disease.21 Although it remains unclear to what degree a single PINK1 mutation is a risk factor for parkinsonism, our findings support the possibility.
Acknowledgments
We thank the patients who participated in this study. We also thank Mary Hulihan, Sarah Lincoln and Minnie Schreiber for technical assistance.
REFERENCES
Footnotes
-
Funding: This study was supported by the Research Council of Norway (grant 153487/V50), the Udall Center at Mayo Clinic Jacksonville (NINDS NS40256), Reberg’s legacy and the Norwegian Parkinson Foundation.
-
Competing interests: None declared.