Article Text

Abstract
The case of a male infant with neonatal encephalopathy and a de novo MECP2 mutation is reported. The presenting phenotype of decelerating head growth, spasticity, scoliosis, and central respiratory disturbance raised suspicions of the diagnosis. Neonatal encephalopathy in males is part of a spectrum of disorders caused by MECP2 dysfunction.
- de novo mutation
- Rett syndrome
- X chromosome inactivation
- recurrence risk
Statistics from Altmetric.com
Mutations in the gene for the transcriptional repressor, MECP2, have been well characterised in girls with Rett syndrome.1 MECP2 mutations are now associated with a widening range of clinical presentations, including Angelman syndrome and X linked and non-specific mental retardation.2–4 There are also reports of males with fatal and non-fatal neonatal encephalopathy caused by an MECP2 mutation identified because of recurrence among siblings or the occurrence of a close female relative with Rett syndrome.5–8 This is the first report of fatal neonatal encephalopathy caused by MECP2 dysfunction, diagnosed in the absence of any supporting family history.
CASE REPORT
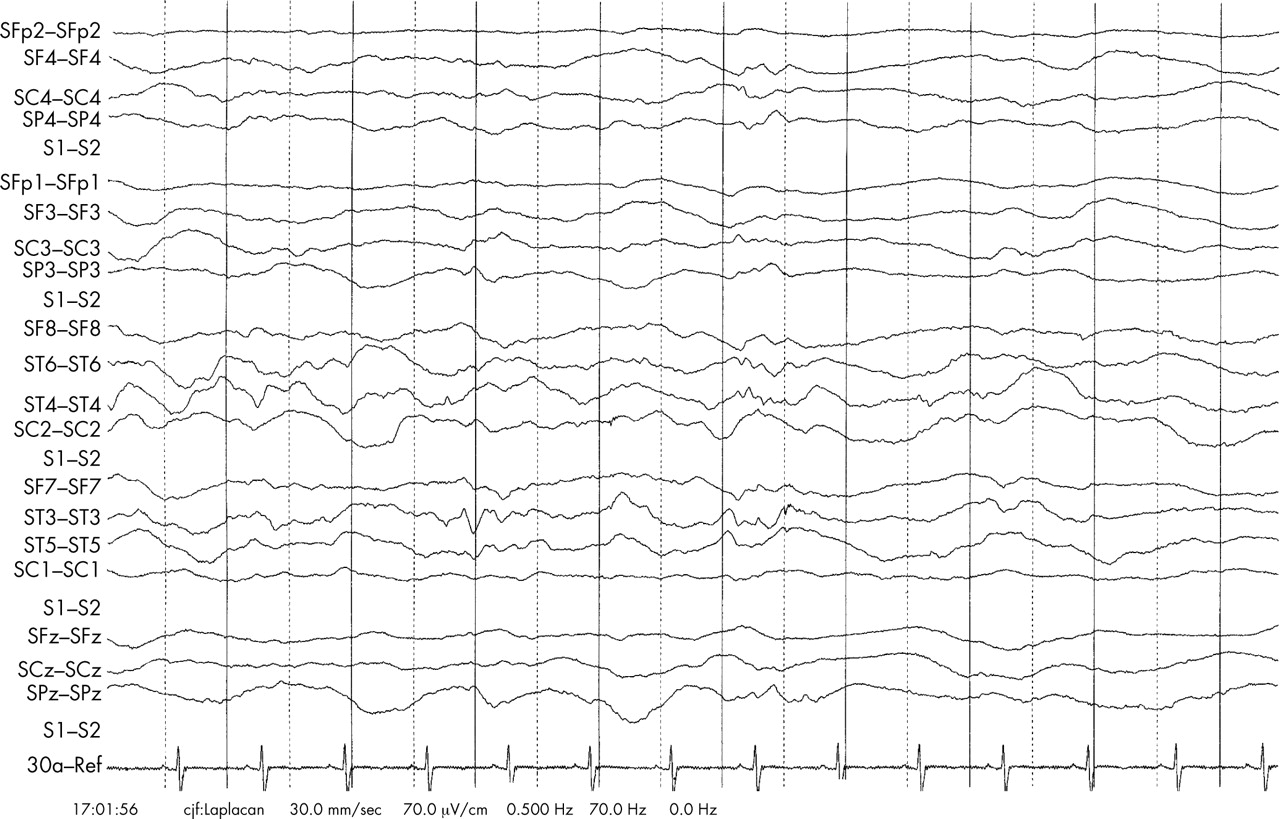
A full term, male child weighing 4000 g was born to non-consanguineous parents after an uneventful pregnancy. Although in good condition immediately after birth, he then fed poorly and was admitted to the special care baby unit at 14 hours of age. Ophisthotonic posturing, abnormal cycling movements of the legs, and increased muscle tone were observed together with pronounced variation in cardiac and respiratory rates. Ventilatory support was not required. An electroencephalogram showed diffuse abnormality but no epileptiform discharges. The results of a full metabolic work up and neuroimaging (magnetic resonance) were within normal limits. He was discharged home aged 2.5 weeks, although he required tube feeding for three months. Repeated paediatric assessment documented neurodevelopmental regression, particularly of motor skills, and the emergence of a scoliosis. His head growth decelerated. At 13 months, head circumference was 46 cm (between the 2nd and 9th centiles), in contrast with being on the 90th centile at birth (36 cm). Repeat magnetic resonance neuroimaging showed increased extra-axial spaces, indicating the development of cerebral atrophy. At 14 months of age, he presented in an obtunded state, apnoeic and cyanosed. Seizures were noted after intubation. Examination found persistent choreiform movements, limb spasticity, and weak respiratory effort. After stabilisation and removal of sedation, he remained ventilator dependent because of severely impaired central respiratory drive. An electroencephalogram showed only diffuse encephalopathy (fig 1). In view of his poor prognosis, his parents permitted extubation and he died within 36 hours.
{kind=link}
Electroencephalogram taken after admission at 14 months of age. The patient was ventilated and was receiving sodium valproate, trimeprazine, atracurium, and chloral hydrate. The predominant abnormality is a diffuse encephalopathy.
MOLECULAR ANALYSIS
MECP2 gene sequencing showed a single nucleotide substitution in exon 4 (c.473C>T), predicted to cause a missense mutation (T158M) within the methylcytosine binding domain of the methyl-CpG binding protein. No evidence of this was present in maternal DNA derived from blood.
DISCUSSION
The specific clinical features that raised suspicion of MECP2 dysfunction were the combination of male neonatal encephalopathy, decelerating head growth, spasticity, scoliosis, and central respiratory disturbance.
The T158M missense mutation characterised is a common recurrent mutation in Rett syndrome. Interestingly, it has been reported in a sibship, comprising a girl with Rett syndrome and her two brothers who had fatal neonatal encephalopathy.6 Their healthy mother also had the mutation but was shown to have skewed X chromosome inactivation (ratio 99:1). In X linked disorders in which the phenotype in females may be modulated stochastically by X inactivation, the male phenotype provides a more accurate representation of the true biological effect of a particular mutation. The severe clinical course of these male cases provides strong empiric support for MECP2 functional studies, which show that the T158M mutation is associated with loss of MECP2 selectivity for methylated DNA.9
Until recently, the paucity of recorded cases of males with MECP2 mutations was attributed to Rett syndrome being an X linked dominant disorder with male lethality. It is now appreciated that most MECP2 mutations occur on the paternally inherited chromosome and that the rarity of maternally derived mutations is responsible for the scarcity of affected males.10 Although rare, it is likely that males with MECP2 mutations are not being recognised because they seldom present with a classical Rett phenotype, the current diagnostic focus for MECP2 mutation analysis. A similar severe phenotype may occur in females as a result of unfavourable skewing of X chromosome inactivation, although this is yet to be reported.
A mutation on the maternally inherited X chromosome may occur in a number of ways: (a) after fertilisation, with resultant somatic mosaicism in the child and a milder phenotype; (b) as a de novo mutation in the ovum; (c) maternal gonadal mosaicism;(d) inheritance of a maternal germline mutation, which will be present in all maternal cells but masked by favourably skewed X chromosome inactivation. The previously published reports of males with fatal neonatal encephalopathy and a characterised mutation have been from families ascertained because of the co-occurrence of a case of Rett syndrome or recurrence of fatal neonatal encephalopathy. In each of these situations, there was either a maternal germline mutation associated with skewed X chromosome inactivation or maternal gonadal mosaicism.5–8 The incidence of Rett syndrome is 1 in 10 000–15 000, with almost all cases being due to a paternal mutation (ratio 8:1).10 The incidence of male neonatal encephalopathy is therefore probably the order of 1 in 80 000 (including all four possible mechanisms listed above). Assuming a male livebirth rate of 350 000 annually in the United Kingdom, we would estimate that four to five cases of male encephalopathy result from MECP2 mutations each year in this country. We would estimate the incidence of female neonatal encephalopathy to be in the region of 1 in 400 000 (1 in 10 000 multiplied by 1 in 40 (likelihood of unfavourable skewing)). However, this may be a gross underestimate. Further work, looking prospectively at a large population based cohort of males and females with encephalopathy is required to answer this question. Not only would this establish a true incidence figure, but it would also delineate further the phenotypic differences between an MECP2 encephalopathy from other types. This would also allow best practice guidelines on the investigation of children with encephalopathy to be established in the neonatal units.
The mother in our case did not carry the mutation in her blood. Although this does not exclude maternal germline mosaicism, it is apparent that the risk of recurrence in our case must be lower than in previously reported cases. She has subsequently had another unaffected male pregnancy.
An MECP2 mutation occurring on a maternally inherited chromosome has a higher risk of recurrence than one arising on a paternal chromosome. However, it is evident that the clinical recurrence risk may be influenced greatly by the manner in which cases are ascertained. Screening for a maternal germline mutation is strongly recommended after characterisation of an MECP2 mutation in any male.
Acknowledgments
We thank Angus Clarke, Andrew Kornberg, Neil Stoodley, Alex Murray, and George Elder for their support and helpful observations.