Article Text

Abstract
AIMS To elucidate the molecular genetic defect of X linked congenital nystagmus associated with macular hypoplasia in three white males of a three generation family with clear features of ocular albinism in only one of them.
METHODS A three generation family with congenital nystagmus following X linked inheritance, and associated with macular hypoplasia was clinically examined (three males and two obligate carriers). Flash VEP was performed to look for albino misrouting. DNA samples were subjected to PCR and subsequent analysis using SSCP for all exons of theOA1 gene. RT-PCR was performed on a mRNA preparation from a naevus from one patient. PCR products presenting divergent banding patterns in SSCP and from the RT-PCR were sequenced directly using cycle sequencing with fluorescent chain termination nucleotides and electrophoresis in a capillary sequencer.
RESULTS The index case (patient 1, IV.1) was diagnosed with X linked OA1 at the age of 3 months because of typical clinical features: congenital nystagmus, iris translucency, macular hypoplasia, fundus hypopigmentation, normal pigmentation of skin and hair, and typical carrier signs of OA1 in his mother and maternal grandmother. Pigmentation of the iris and fundus had increased at the last examination at age 4 years. Albino misrouting was present at this age. In the maternal uncle (III.3, 51 years) who also suffered from congenital nystagmus there was clear macular hypoplasia and stromal focal hypopigmentation of the iris but no iris translucency or fundus hypopigmentation. Patient 3 (II.3, 79 years, maternal uncle of patient III.3) had congenital nystagmus and was highly myopic. The fundus appearance was typical for excessive myopia including macular changes. The iris did not show any translucency. Molecular genetic analysis revealed a novel 14 bp deletion of theOA1 gene at nt816 in exon 6. The mutation abolishes four amino acids (Leu 253-Ile-Ile-Cys) and covers the splice site. Nucleotides 814/815 are used as a new splice donor thus producing a frame shift in codon 252 and a new stop codon at codon 259.
CONCLUSIONS Macular hypoplasia without clinically detectable hypopigmentation as the only sign of X linked OA1 has been reported occasionally in African-American, Japanese, and white patients. The present family shows absent hypopigmentation in two patients of a white family with a deletion in the OA1 gene. We propose a model of OA1 that allows increase of pigmentation with age. We hypothesise that macular hypoplasia in all forms of albinism depends on the extracellular DOPA level during embryogenesis, and that in OA1 postnatal normalisation of the extracellular DOPA level due to delayed distribution and membrane budding/fusion of melanosomes in melanocytes results in increasing pigmentation.
- ocular albinism
- OA1 gene
- congenital X linked nystagmus
Statistics from Altmetric.com
Congenital nystagmus (CN) is the common symptom of a range of diseases involving the macula from infancy on. Diagnosis of the underlying disease often requires extensive clinical, electrophysiological, psychophysical, and eventually molecular genetic examinations, especially when clinical findings are unrevealing.1
One disease with CN is ocular albinism. X linked ocular albinism (OA1, OMIM: 300500) results from a defect in the cellular distribution of melanin in the retinal pigment epithelium (RPE) and iris pigment epithelium (IPE), and also in melanocytes and keratinocytes of the choroid and skin. The phenotype is caused by mutations in theOA1 gene.2 The locus was assigned to the distal short arm of the X chromosome at Xp22.3–22.2. The OA1 gene product is a glycoprotein in the membrane of melanosomes which binds trimeric G proteins and has currently been shown to be involved in intracellular signal transduction in the lysosomal/melanosomal sorting pathway.3
The most prominent findings in hemizygous males with OA1 are iris translucency, macular hypoplasia, and fundus hypopigmentation together with normally pigmented skin and hair.4 Macromelanosomes in the pigment epithelium5 of the eye and skin are a typical but not specific finding in hemizygous males and carrier females, and indicate that the pathological process is not limited to the eye. Affected males show strabismus, marked photophobia and, as in other types of albinism, misrouting of the optic tracts.6Female carriers present varying degrees of iris and fundus hypopigmentation in a high proportion of cases.7
We present a three generation family with CN following an X linked inheritance pattern, and associated with macular hypoplasia in three white males. Molecular genetic analysis of theOA1 gene was performed since one of the patients (IV.1) showed clear features of ocular albinism. We characterised the underlying molecular defect as a novel 14 bp deletion (816del14bp) in the OA1 gene causing a frame shift due to aberrant splicing.
Methods
CLINICAL EXAMINATION
A three generation family with CN following an X linked inheritance pattern, and associated with macular hypoplasia (Fig 1) was examined clinically including slit lamp biomicroscopy and indirect ophthalmoscopy (three males and two obligate carriers). The anterior and posterior segments of the eyes were documented by slit lamp (Zeiss, Jena) and fundus photography (Topcon, Willich). In one affected male (IV.1), flash VEP was performed at age 4 years (Spirit Nicolet, Madison, WI, USA; flash intensity 2 cd/m2.s, reference FZ, registration O1= 4 cm left to CZ, O2 = 4 cm right to CZ, CZ 2 cm above inion).

Pedigree of family 74 segregating the OA1 gene mutation.
SKIN BIOPSY
From patient III.3 a skin biopsy of a naevus was taken at the department of dermatology to obtain melanocytes for RNA studies, and for histological examination to detect macromelanosomes.
MOLECULAR GENETIC STUDIES
DNA was extracted from peripheral blood lymphocytes according to a previously reported method.8
DNA samples were subjected to PCR using oligonucleotide primers as reported by Schiaffino et al.9All coding exons of the OA1 gene including 85 bp of the 5' UTR and 336 bp of the 3' UTR were amplified.
Subsequent analysis of the PCR products was performed using single strand conformation polymorphism analysis (SSCP). SSCP analysis was done on PAGE gels (11 cm) using 10% polyacrylamide (19:1) with a 5 mm overlay of 6% polyacrylamide (19:1) in 1x TBE buffer. The gels were run for 12–14 hours at 140–180 V constant and 10°C.
PCR products presenting divergent banding patterns in SSCP as well as the PCR products from RT-PCR were sequenced directly using cycle sequencing with fluorescent chain termination nucleotides and electrophoresis in an ABI 310 capillary sequencer.
To confirm the sequencing result the exon 6 PCR products of the patients and controls were tested for the loss of a Tsp509I restriction site.
The effects of the identified mutation on RNA splicing were studied by PCR amplification of the mRNA of the OA1gene from exon 4 to exon 7. Therefore, mRNA was isolated from naevus melanocytes of patient III.3. Control RNAs were prepared from retina, retinal pigment epithelium (RPE), and choroid + RPE from a donor eye of an unaffected individual and a cDNA preparation from melanocytes kindly provided by B Becker, Department of Dermatology, University of Regensburg, Germany.
We designed primers (OA4-7a: 5′-CCGC CATGCTCTACTACCCTTC-3′; OA4-7b: 5′-TGGCTGCAGTTCTGACAGGTTT-3′) based on Genbank entry NM_000273 to perform the RT-PCR. The calculated size of the RT-PCR product was 349 bp.
Results
CLINICAL DATA
The pedigree of family 74 segregating CN is shown in Figure 1.
PATIENT 1 (IV.1)
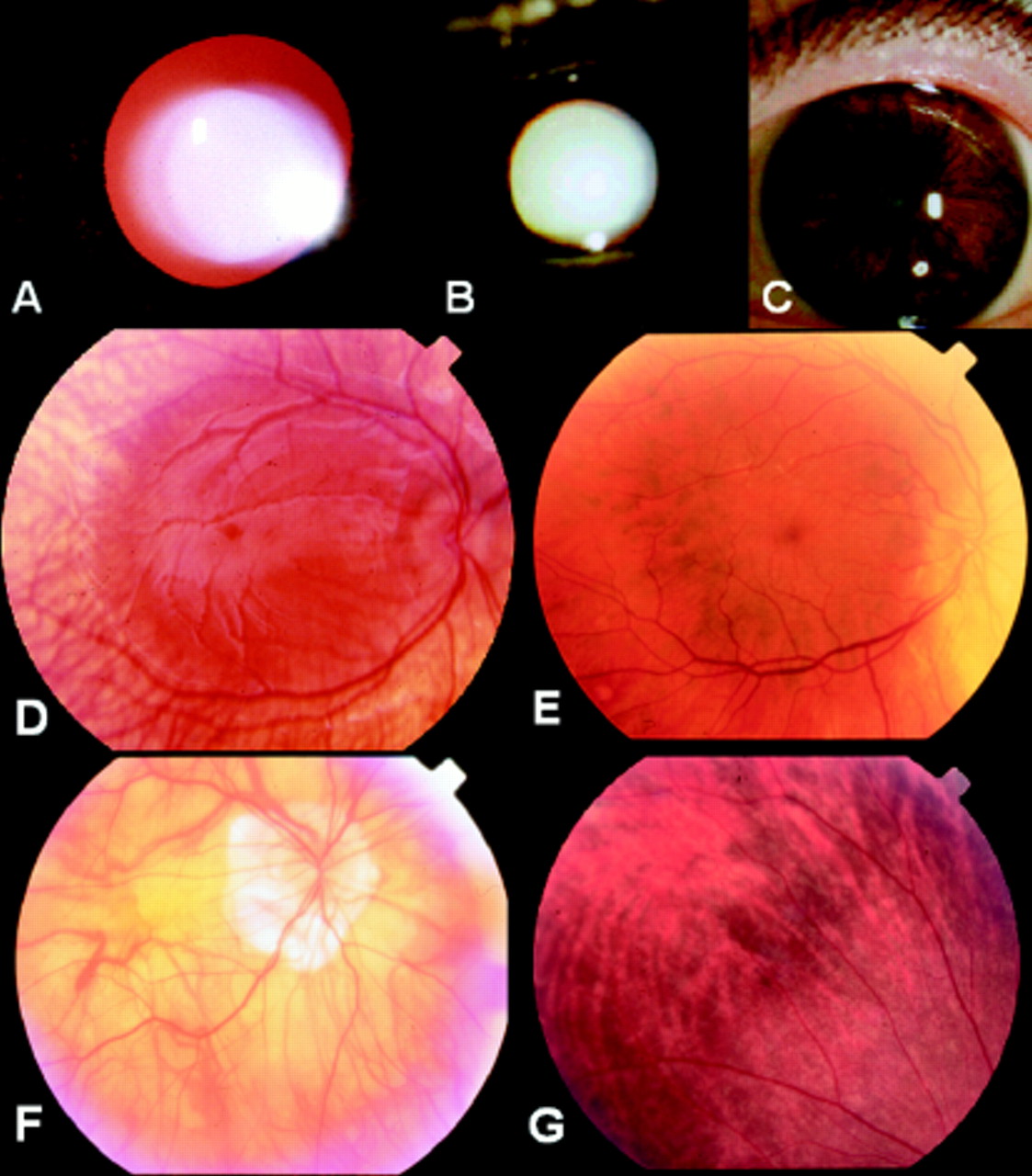
The index patient was diagnosed with OA1 at age 3 months because of typical clinical features: CN, iris translucency, macular hypoplasia, fundus hypopigmentation, normal pigmentation of skin and hair (Fig 2A), and typical carrier signs of OA1 in his mother and maternal grandmother (see below). At age 2.5 years VA was 0.12 bin (Lea test, sc). At age 4 years his visual acuity had improved to 0.4 Lea test bin (RE plano/−2.5 cyl A 170°, LE plano/−2.5 cyl A 10°). Fundus and iris pigmentation had also increased (Fig 3A and D). Flash albino VEP showed clear asymmetry—that is, much smaller amplitudes on the left occipital hemisphere when stimulating the right eye.
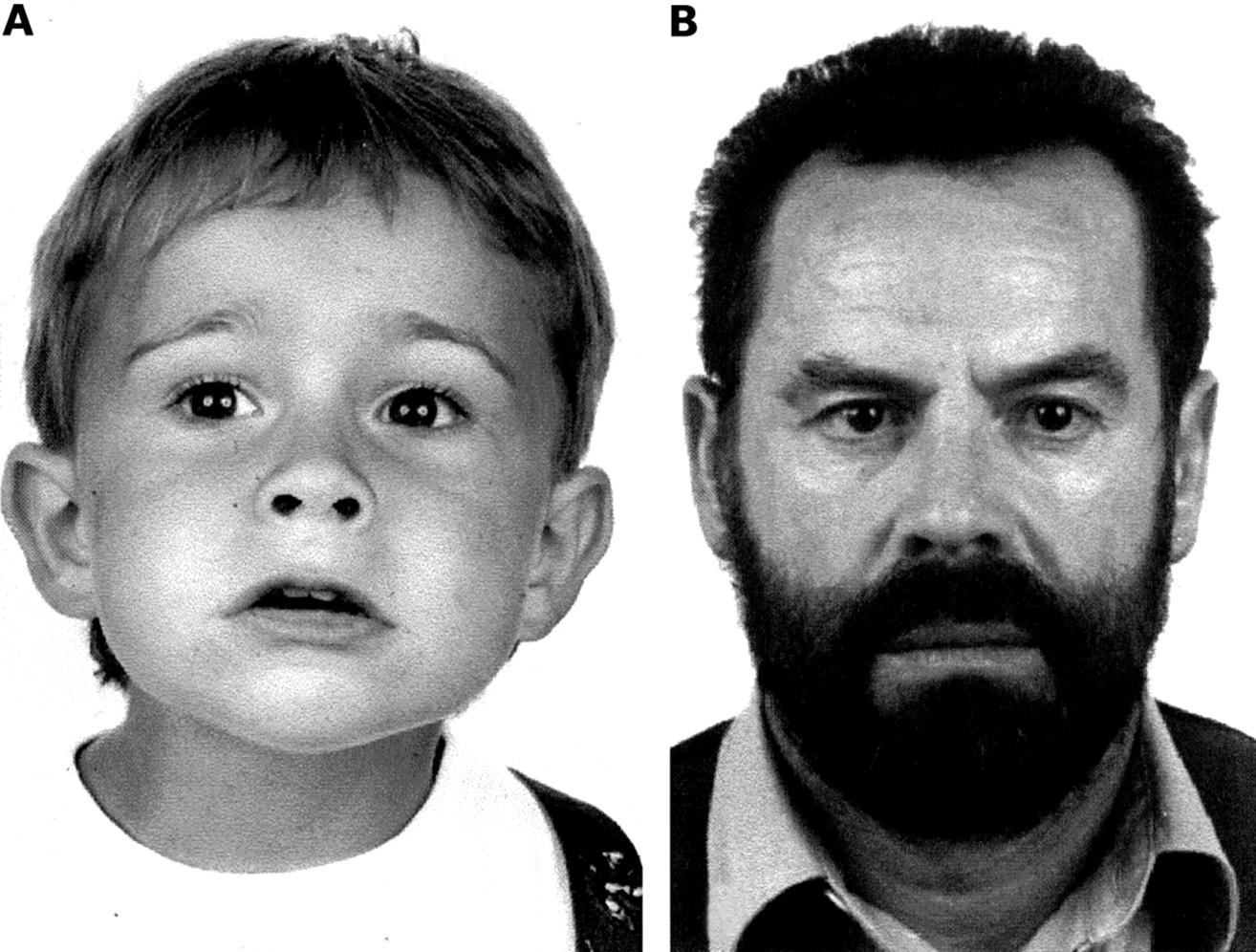
External aspect of (A) patient IV.1 at age 2.5 years and (B) patient III.3 at age 54 years. Tanning is obvious in both patients. Patient III.3 additionally has very dark hair colour.

Testing of iris translucency in patient (A) IV.1 and (B) III.3, and iris pigmentation in patient III.3 (C). Patchy hypopigmentation can be seen in the iris stroma of patient III.3 but no obvious translucency can be detected compared with patient IV.1. Fundus appearance showing macular hypoplasia and varying degrees of hypopigmentation in patients IV.1 (D), III.3 (E), II.3 (F), and the carrier III.2 (G) (mother of patient IV.1).
PATIENT 2 (III.3)
In the maternal uncle of patient IV.1 aged 51 years there were clear macular hypoplasia (Fig 3E), patches of hypopigmentation of the iris stroma (Fig 3C) but no iris translucency (Fig 3B) or fundus hypopigmentation (Fig 3E). It is interesting to note that both hair and iris stroma (outside the hypopigmented patches) were heavily pigmented (Fig 2B, Fig 3C). His VA was 20/200 in both eyes (RE −2.75 sph/−4.75 cyl A 35°, LE −3.0 sph/−4.25 cyl A 170°). Nystagmus had been present since infancy.
Histological examination of a naevus biopsy revealed macromelanosomes.
PATIENT 3 (II.3)
The maternal uncle of patient III.3 reported to have CN since early childhood. He was originally highly myopic (−18 dioptres in both eyes). At age 78 years, he had bilateral posterior chamber IOL implantation. At presentation he was 79 years old. His VA was then 20/200 in both eyes (RE +2.0 sph, LE 2.0 sph). The iris did not show any translucency. The fundus appearance was typical for excessive myopia including macular changes (Fig 3F). Nystagmus was still present at examination.
CARRIER III.2
Her visual acuity was normal (RE −3.5 sph = 0.9, LE −3.5 sph = 1.0) at age 37 years. There was no iris translucency but hypopigmented patches of the iris stroma in both eyes similar to those in her brother III.3. On funduscopy, there was a typical mosaic pattern of hypopigmented RPE intermixed with normally pigmented RPE (Fig 3G).
CARRIER II.2
At examination she was 76 years old. She was bilaterally aphakic. Her VA was 0.8 in both eyes (RE +12.5 sph, LE + 13.25 sph/−4.0 cyl A 75°). She had clear iris translucency, and on funduscopy there was a typical mosaic pattern of hypopigmented RPE intermixed with normal pigmented RPE similar to that in her daughter (III.2).
MOLECULAR GENETIC ANALYSIS
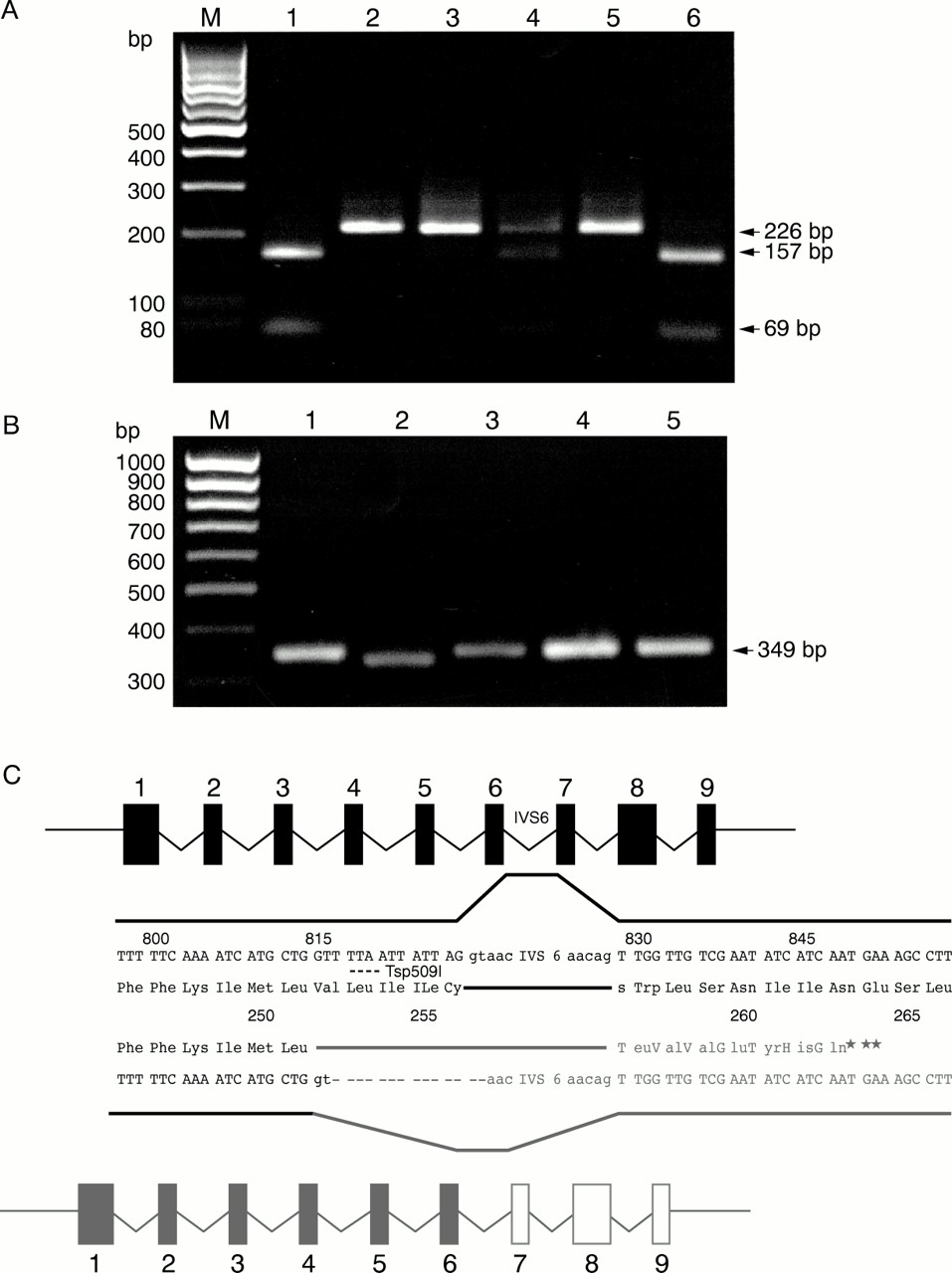
SSCP analysis showed an aberrant banding pattern for the exon 6 amplimer of the OA1 gene. Direct sequencing of the PCR product revealed a 14 bp deletion starting at nt816 and covering the splice donor signal (gt) (Fig 4C). (Nucleotide numbering based on the cDNA reported by Bassi et al,2 Genbank: NM_000273.) The mutation abolishes a restriction enzyme recognition site for Tsp509I. A loss of the restriction site could be shown in all patients. The heterozygous carrier status could be shown in the mother of patient IV.1 (Fig4A).

(A) Tsp509I restriction enzyme digest. The wild type sequence is cut once to produce fragments of 157 bp and 69 bp. This site is lost by the 816del14bp mutation. Lanes: (1) control, (2) patient II.3, (3) patient IV.1, (4) carrier III.2, (5) patient III.3, (6) control. (M) Marker: 100 bp ladder. (B) RT-PCR using RNA from (1) control melanocytes, (2) patient III.3 melanocytes, (3–5) preparations from a control donor eye (3) retina, (4) RPE and choroid, (5) RPE with choroid contamination. (M) Marker: 100 bp ladder. (C) Schematic showing the effect of the 816del14bp mutation on RNA splicing. The mutant gene is shown in grey.
FUNCTIONAL PROOF
Since the mutation covers the splice donor of intron 6, we assumed an effect on splicing, as the flanking sequence provides enough information to produce an alternative splice site (Fig 4C).
To evaluate this splice site RT-PCR was performed to amplify a product of 349 bp (Fig 4B). A product of the expected size could be detected in a cDNA preparation of RNA from melanocytes (lane 1), retina, RPE, and choroid + RPE (lanes 3–5) from control individuals. RNA of melanocytes from a naevus of patient III.3 showed a PCR product of reduced size (lane 2). Direct sequencing of the RT-PCR product of patient III.3 and a control sample revealed a mRNA which used nucleotides 814–815GT as splice donor signal (Fig 4C) in the RNA of patient III.3. Therefore, the open reading frame (ORF) of the resulting mRNA comes into a frame shift predicting the termination of the resulting gene product at amino acid 259 (Fig 4C).
Discussion
We report on a novel mutation (816del14bp) in theOA1 gene.
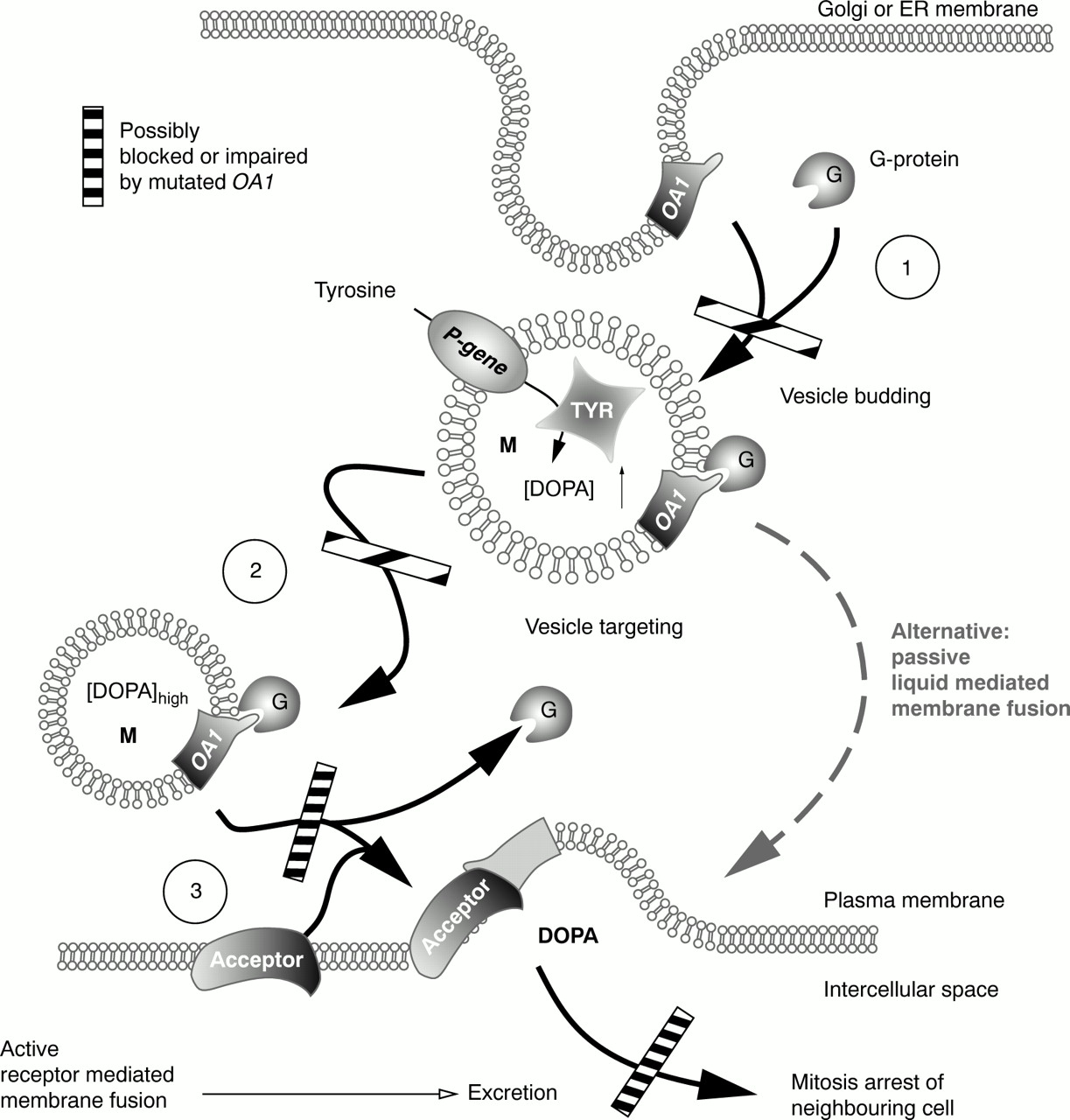
OA1 codes for a seven helix transmembrane G protein coupled receptor (GPCR) of melanosomal membranes. GPCRs transduce signals over membranes and activate second messenger signalling cascades by coupling to GTP binding proteins. The function of OA1 is not defined currently. It may represent a receptor for a G protein which is involved in regulating the biogenesis and maturation of melanosomes (Fig 5(1) and (2)).3
OA1 patients present macromelanosomes as a typical feature. Garneret al10 hypothesised macromelanosomes to result from promelanosomes which do not separate correctly from the endoplasmatic reticulum and Golgi apparatus. This notion is supported by a recent animal model of Oa1knockout mice.11 Ultrastructural analysis of the RPE of these mice showed normal melanosomes from E12.5 until birth. Macromelanosomes started to form at P1. Individual macromelanosomes were absent in adult skin, hair and uveal melanocytes11which showed darker pigmentation than the RPE.
We conclude that mutated OA1 causes late budding and may lead to increased melanosome size—that is, macromelanosomes which accumulate at the site of their origin owing to impaired budding signals. The same defect may impair active fusion of melanosomes and the plasma membrane (Fig 5(3)) resulting in a transient hypopigmentation as seen in patient IV.1.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic indicating maturation steps of melanosomes possibly impaired by mutated OA1. Budding of vesicles from ER and Golgi membranes is controlled by small G proteins (Ras or Rab) which may represent ligands to the GPCR OA1. Mutations in OA1 would cause a delay in budding and, therefore, vesicles with increased size. (2) The same group of G proteins controls targeting of vesicles (melanosomes) to intracellular compartments and the plasma membrane. Mutations in OA1 would cause intracellular misrouting of vesicles. (3) As a third function of a transmembrane GPCR, interaction of OA1 with an acceptor molecule on the plasma membrane can be proposed. Absent or altered OA1 would cause impaired DOPA excretion by impaired membrane fusion. An alternative way of excretion would be passive lipid mediated membrane fusion which rarely occurs spontaneously at membrane surfaces but requires more time. Any of the steps mentioned above cause impaired excretion of DOPA and even if passive membrane fusion or vesicle targeting occurs, it will not be possible to excrete sufficient amounts of DOPA in time.
The mutation in the OA1 gene reported here affects the splice donor site of intron 6 including the complete splice signal. An influence on splicing could be shown in patient III.3 (who showed the less severe phenotype as to ocular hypopigmentation). In a preparation of cDNA from his dermal melanocytes a PCR product of the OA1 mRNA was detectable but showed an aberrant size. Since the splice donor signal was deleted nucleotides 814/816 were used as a new splice donor signal. The newly created splice signal has a quality equal to the optimal splice signal as reported by Shapiro et al.12The resulting mRNA showed a functional frame shift abolishing the correct translation of the mRNA after Leu251 and predicting the termination at codon 259. The resulting gene product lacks about one third of the amino acids including the last transmembrane domain from the extracellular loop 3 up to the C terminus.
Further examples of GPCRs relevant in the eye are rod and cone visual pigments which perceive light and activate the G protein transducin to start a second messenger cascade. Mutations in these are involved in retinal disorders. Rhodopsin interacts with the α subunit of transducin at its fourth cytoplasmic loop.13 Deletions downstream of the third extracellular loop of rhodopsin cause ADRP.1415 We therefore assume that this novel hypomorphic OA1 gene mutation impairs the interaction of OA1 with the associated G protein, thus abolishing vesicle budding (Fig 5(1)), vesicle targeting (Fig 5(2)), or active membrane fusion of melanosomes and the plasma membrane (Fig 5(3)).14
Melanosomes generate melanin from tyrosine via 3,4-dihydroxyphenylalanine (DOPA). DOPA concentration is reduced when tyrosinase activity (oculocutaneous albinism, OCA1) is reduced, or when reduced P-gene activity abolishes supply of tyrosine (OCA2). DOPA is an antimitogenic factor involved in cell cycle regulation and crucial for the maturation of the retina and the optic nerve.16 The neuroretinal pathology of OCA is caused by the lack of DOPA, thus giving an overdevelopment of retinal cells and cells of the optic nerve.16 This results in a developmental injury during embryogenesis causing a neuroretinal phenotype which cannot be reversed during postnatal development. In OA1, the studies summarised above imply that melanosomes do not actively excrete the formed DOPA, thus not reaching a DOPA concentration critical for the development of the neuroretina in due course—that is, during embryogenesis. After birth neuroretinal cells will cease to proliferate. Therefore, the neuroretinal phenotype is permanent in all three forms of albinism even if there is increasing pigmentation postnatally. In OA1 melanocytes form melanin throughout life in contrast with OCA. This will increase pigmentation over time in the iridal pigment epithelium (IPE), RPE, and skin, and also in the uveal melanocytes.11 Marked postnatal activity could be shown in the iridal stroma of cynomolgus monkeys after induction by latanoprost,17 thus explaining the decrease of iris translucency in our youngest patient. The variable degree of hypopigmentation in OA1 patients may be explained by the complex interaction of the genes involved in melanin synthesis.
Our hypothesis on the pathogenesis of the phenotype associated withOA1 mutations is additionally supported by other occasional reports of macular hypoplasia without clinically detectable hypopigmentation as the only sign of OA1 in African-American, Japanese, and also one white patient.18-22 It shows that visual acuity may be greatly impaired even in the absence of macular hypopigmentation although from literature data it is evident that there is a trend towards better visual acuity with increasing pigmentation.20 The lack of hypopigmentation may impede the recognition of OA1 in some older patients, and can only be overcome by carefully searching for additional signs such as macular hypoplasia and albino misrouting.
Acknowledgments
The authors wish to thank W Stolz and B Becker from the department of dermatology, University of Regensburg for their helpful assistance in naevus biopsy of patient III.3 and providing control samples of RNA from melanocytes. We also wish to thank I Russel-Eggitt, London, for asking a very helpful question.
This work was supported by the Deutsche Forschungsgemeinschaft (Lo457/3-1,2).