Article Text

Abstract
Aim: To phenotype and genetically map the disease locus in a family presenting with autosomal dominant microcornea, rod-cone dystrophy, cataract, and posterior staphyloma.
Methods: Six affected and three unaffected members of the pedigree were examined. All individuals provided a history and underwent a full clinical examination with A-scan and B-scan ultrasonography and electrophysiological testing where appropriate. PCR based microsatellite marker genotyping using a positional candidate gene approach was then performed on DNA samples extracted from venous blood provided by each subject.
Results: The disorder is inherited as an autosomal dominant trait with variable expressivity and has a complex phenotype. Affected individuals had bilateral microcornea, pulverulent-like lens opacities, a rod-cone dystrophy and posterior staphyloma (MRCS). Using a positional candidate gene approach, the authors have evidence suggestive of linkage of this disorder to a region on 11q13 within the nanophthalmos 1 (NNO1) genetic interval. The small family size militates against achieving a LOD score of 3, but the haplotype data and the position of the putative MRCS locus within a known nanophthalmos locus are suggestive of linkage. A candidate gene within this region (ROM1) was screened and no mutations were found in affected members of the family.
Conclusion: This rare developmental disorder has some phenotypic similarities to nanophthalmos and possibly maps to a locus within the genetic interval encompassing the NNO1 locus. Screening of candidate genes within this region continues.
- molecular genetic study
- MRCS
- microcornea
- rod-cone dystrophy
- cataract
- posterior staphyloma
Statistics from Altmetric.com
Eye development is directed by the coordinated temporal and spatial interaction of a number of genes and their products. A mutation in any one of these developmental genes therefore would be predicted to manifest as a complex ocular phenotype affecting multiple structures within the eye. An increasing number of genes have been identified as causing various forms of anterior segment dysgenesis and abnormalities of ocular size. Some—for example PAX61, CHX10,2 and SIX3,3 are associated with small eyes and others, PITX2,4PITX3,5FOXC1,6FOXE3,7PAX6,8 EYA1,9MAF,10 and CYP1B1,11 are associated with anterior segment dysgenesis.
A number of clinical disorders affecting development of the globe are recognised. Anophthalmia is a term used to describe complete absence of the globe; this is however a histological diagnosis and rare as small remnants of ocular tissue are usually present within the orbit.12 The term clinical anophthalmos is more appropriate to describe patients who appear to have no ocular structures on clinical examination. In microphthalmos, the eye is small13 and there is often associated ocular coloboma.14 Anophthalmos and microphthalmos probably form part of the same disease spectrum and both may be seen within the same family.15 Asymmetrical or strictly unilateral involvement may be seen. Both may be associated with other systemic abnormalities.
Nanophthalmos, described by some authors as “simple microphthalmia,”12 is an uncommon usually bilateral ocular condition characterised by a small eye with (a) a reduced axial length, (b) high hyperopia16 (+7.00 D to + 13.00 D), (c) a high lens/eye volume ratio, (d) a high incidence of angle closure glaucoma,17 and (e) characteristic yellow macular pigmentation, chorioretinal folds, and crowded optic discs.18 Ocular ultrasound commonly reveals a thickened sclera composed of abnormal collagen fibrils19 which may obstruct the outflow of blood through the vortex veins resulting in recurrent or persistent choroidal effusions16 and non-rhegmatogenous retinal detachments. Rarely, nanophthalmos has been reported in association with a pigmentary retinopathy.20–22
Nanophthalmia is usually sporadic but may be inherited as an autosomal dominant or recessive trait. It can also be seen in association with other systemic congenital abnormalities.13 The genetic locus for the recessive form of nanophthalmos has not been identified but linkage analysis of a single dominant family has assigned a locus (NNO1)23 from 11p12 to 11q13 (an interval close to but excluding the PAX6 gene1).
In this paper, we report the findings in a three generation English family with an autosomal dominant ocular phenotype comprising microcornea, rod cone dystrophy, cataract, and posterior staphyloma (MRCS) possibly mapping genetically within the NNO1 locus on chromosome 11. The implications of this observation are discussed.
SUBJECTS AND METHODS
Subjects
The clinical genetic database at Birmingham Women’s Hospital, Birmingham, UK, was used to identify a family, the affected members of which exhibited a complex developmental disorder of the eye. Eleven members of the family agreed to take part in the study (Fig 1). The study had the approval of the Moorfields Eye Hospital ethics committee. Informed consent for clinical and genetic investigations was obtained from all participants.

Family pedigree and haplotype analysis using microsatellite markers on 11q13. Affected individuals are denoted by solid symbols.
Clinical examination
An ophthalmic and general medical history was obtained and each patient underwent a systemic and full ophthalmic examination. The ophthalmic examination included assessment of visual acuity and detailed examination of the anterior and posterior segments and tonometry. Five individuals underwent electroretinography and two had electro-oculography. Older subjects had electroretinography performed according to ISCEV protocol,24 but the children who were unable to tolerate gold foil electrodes had electroretinograms performed with the skin electrodes. Six affected individuals also had A-scans and B-scan ultrasonography.
Genotyping
Genomic DNA was extracted from EDTA sequestered blood samples taken using the Nucleon II DNA extraction kit (Scotlab Bioscience). PCR based genotyping using microsatellite markers (dinucleotides, trinucleotides, and tetranucleotides) were used. The amplified PCR products were resolved by 6% non-denaturing polyacrylamide gel electrophoresis (Protogel; National Diagnostics, Atlanta, GA, USA) and were visualised by staining with ethidium bromide. Subsequent genotyping and haplotype analysis were performed manually. We used a positional candidate gene approach to determine whether loci associated with small eyes or nanophthalmia were responsible for this unusual syndrome. Candidate loci were located on the long arm of chromosome 14 (CMIC25and CHX102), chromosome 11 (NNO123 and PAX61) and the long arm of chromosome 15 (NNO226). Pedigree data were collated with pedigree mapping software (version 2.1.3; Cherwell Scientific Publishing Ltd, The Magdalen Centre, Oxford Science Park, Oxford, UK) and two point linkage analysis performed using the MLINK component of the linkage program, version 5.127 using an autosomal dominant model, equal allele frequencies, a gene frequency of 10−4, and a mutation rate of 10−6.
Mutation screening
Mutation screening of a possible candidate gene, ROM1, within this region was performed. Coding exons and flanking splice junctions were screened for mutations by direct sequencing. Reactions were performed in a 50 μl volume with 200 ng of genomic DNA; 20 pmol of each primer; 200 μM each dATP, dCTP, dTTP, dGTP; 10 mM TRIS-HCl (pH 8.3); 1.5 mMol MgCl2, and 0.5 U Taq DNA polymerase (Bioline Biotaq). After the initial denaturation step at 95°C for 3 minutes, the samples were processed through 30 cycles of 95°C for 30 seconds, 55–60°C for 30 seconds, and 72°C for 45 seconds. A final extension step was performed at 72°C for 5 minutes. After amplification, PCR products were processed using a PCR purification kit (Quickstep PCR Purification Kit) according to the manufacturer’s instructions. A volume of 1.5 μl of the purified PCR product was used in a sequencing reaction. Both the forward and reverse strands of the PCR products were directly sequenced (ABI 373 sequencer, using the ABI prism BigDye Terminator cycle sequencing ready reaction kit; PE applied Biosystems, Foster City, CA, USA). Sequencing reactions were set up according to the manufacturer’s instructions, using either the forward or reverse primers.
RESULTS
Six individuals, three males and three females (age range 9–56 years), were found to be affected. The disorder was inherited as an autosomal dominant trait. Typically patients complained of night blindness during their teenage years and poor vision due to cataracts before the age of 30. Cataract surgery was performed in the second or third decade. One affected member (III:2) was unavailable to attend for a comprehensive assessment, but review of his clinical records revealed the findings of microcornea, a pigmentary retinopathy, and previous cataract surgery. Older individuals had poor vision (ranging from no perception of light to 20/400) at the time of examination while the younger individuals retained good visual acuities (20/30) (Table 1). All individuals had small corneas (Table 1 and Fig 2A and B) and two older individuals (II5 and II7) had chronic angle closure glaucoma. Younger individuals had pulverulent-like cataracts, whereas older individuals were aphakic as a result of previous cataract surgery. The phakic individuals were moderately myopic (Table 1).
Clinical evaluation of the phenotype

(A) and (B) Microcornea in individual II5. (C) Left inferonasal peripheral fundal view in individual IV1 showing demarcation line. (D) The right posterior pole in individual II:7 with arrow depicting the optic disc. (E) Oblique longitudinal ultrasound section demonstrating the posterior staphyloma in the left eye (individual II:5) SUP = superior, INF = inferior, POST = posterior, A = anterior.
All affected members had retinal abnormalities consisting of peripheral retinal pigment epithelium atrophy and retinal pigmentation. In addition, there was evidence of a posterior staphyloma in nine of 12 eyes. The younger individuals showed a clearcut demarcation line, which may be related to the boundary of the staphyloma. Anterior to this line was evidence of retinal pigmentation (Fig 2C). Older individuals displayed no such boundary and pigmentation was seen throughout the posterior pole (Fig 2D) and into the posterior staphyloma.
No systemic disease or abnormality was identified as segregating with the retinal disease. Although clinical examination showed some features consistent with a diagnosis of nanophthalmos, patients did not show consistently reduced axial length. Ultrasonography of nearly all individuals showed abnormally shaped eyes with posterior staphylomata (Fig 2E), but eye sizes within the normal range. The exceptions were individuals III:2 and III:4. III:4 had a posterior staphyloma in one eye and a shortened axial length (without posterior staphyloma) in the other.
The electroretinogram (ERG) was extinguished (Fig 3) in the two older members of the family (average age 54 years). Subnormal photopic and scotopic responses were demonstrated in individuals III:4 and her children IV:1 and IV:2. In Individual IV:1 the scotopic responses were reduced compared to the photopic responses (Fig 3).

Flash ERGs. Left panel: control; middle panel: patient IV:1; right panel: patient II:7. LA = light adapted. DA = dark adapted. SF = standard flash (ISCEV protocol, see text). OPs = oscillatory potentials. The vertical broken line indicates stimulus onset.
Molecular analyses
Markers on 14q excluded the congenital microphthalmia (CMIC) locus and CHX10. Markers on 15q also excluded the NNO2 locus (Table 2). The maximum score for this small family using autosomal dominant inheritance was 2.01 with marker D11S1765 at recombination fraction of zero. A LOD score of 1.98 at 𝛉 =0 was also achieved for marker D11S4076 (0.84 cM telomeric to D11S1765). D11S4191 and D11S1883 were flanking markers with recombinant events, therefore defining a likely 5.0 cM genetic interval within the NNO1 locus. The haplotypes are presented in Figure 1. This region contained several genes one of which, ROM1, is a candidate gene for retinal dystrophy. No sequence change that co-segregated with disease was found in the promoter and coding sequences of this gene.
LOD score showing exclusion data for candidate genes and loci
DISCUSSION
The features of microcornea, rod-cone dystrophy, pulverulent cataract, and posterior staphyloma segregate together in this family and represent a rare ocular phenotype (MRCS). The clinical findings of microcornea and angle closure glaucoma are similar to those found in nanophthalmia but in our family most eyes showed apparently normal axial lengths. Indeed the phakic eyes were myopic (Table 1). However, measurements were complicated by the presence of posterior staphyloma. It is of note that in the affected eyes without posterior staphylomata, the axial lengths were less than 19 mm. Pigmentary retinopathy was seen in all eyes and in the younger members of the family there was a clearcut anterior demarcation line between the normal and dystrophic retina (Fig 2C). Such changes may be seen following resolution of choroidal effusion which may complicate nanophthalmos but the ERG findings of a widespread rod-cone dystrophy in our subjects suggests that the pigmentary change is caused by photoreceptor cell death. Thus, the phenotype may reasonably be considered nanophthalmia plus posterior staphyloma, cataract, and retinal dystrophy. Such an observation is also consistent with our molecular genetic analyses, which suggests that the disease gene maps within the NNO1 locus on chromosome 11.
The association of pulverulent cataract with nanophthalmos has not been previously recorded. The type of opacification, so called because of its dust-like appearance, is one of the most common phenotypes seen in human non-syndromic congenital cataract. Molecular genetic analyses have revealed that mutations within several genes encoding lens specific transmembrane proteins that maintain cell homeostasis28–30 are among the many genes responsible. It is reasonable to suggest therefore that the disease causative gene in our family may encode a similar protein but one which is also expressed in the retina and cornea. Alternatively, the mutation, which, by deduction from the phenotype lies within a gene most likely involved in eye development, might result in downregulation of the expression of one or more of these transmembrane proteins. In keeping with the eye developmental regulator, SIX5,31 this effect may extend to adulthood. The association of a progressive rod-cone dystrophy with developmental structural abnormalities of the eye in our family also supports the notion that the causative gene may have both a developmental function and probably a continuing function after embryogenesis. This dual role is also seen in another developmental gene, PAX6, which gives rise to a developmental ocular abnormality1 and also has a role in maintenance and renewal of the corneal epithelium.32
The association of microcornea/nanophthalmos and pigmentary retinopathy have been reported previously21–23 but a genetic locus has not been identified (Table 3). The spectrum of ocular signs described in other families certainly overlaps the features of the syndrome seen in this family. Notwithstanding, no previously published family has all the components of MRCS. Hermann in 195833 described an autosomal dominant disorder consisting of microphthalmia and pigmentary retinopathy, which has some similarities to the family reported here. The individuals were moderately myopic suggesting, in the presence of microphthalmia, that posterior staphyloma may have been present; axial length measurements were not available at that time. In contrast with our family the expressivity was extremely variable in the French family; of the 19 family members examined, six individuals had microphthalmia and pigmentary retinopathy and seven individuals microphthalmos alone. Although the association of microcornea and posterior staphyloma has been specifically reported,34 no mention of pigmentary retinopathy was made in the published pedigree.
Families and sporadic reports of disorders similar to MRCS
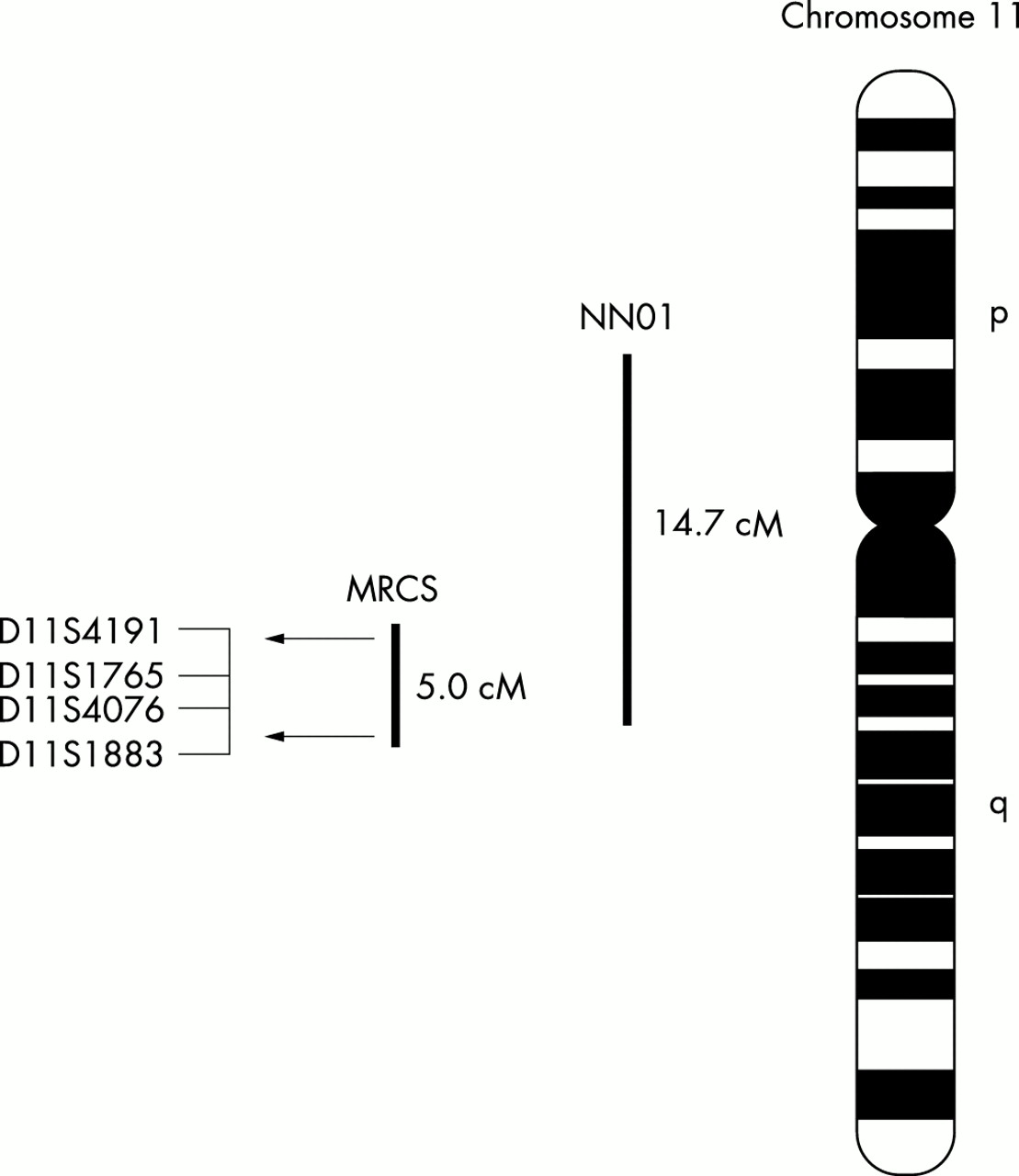
To test the hypothesis that the congenital anomalies observed in our family result from a mutation within an ocular developmental gene, we used microsatellite marker genotyping to exclude genetic linkage in our family to several candidate loci. After excluding linkage to other candidate loci, a positive LOD score (2.01 at 𝛉=0) was obtained for the marker D11S1765 within the NNO1 locus. D11S4076 (a marker which is 0.84 cM telomeric to D11S1765) produced a similarly positive result (1.96 at 𝛉=0). The small family size militates against achieving a LOD score of 3, but the haplotype data and the position of the putative MRCS locus within a known nanophthalmos locus is suggestive of linkage. Nevertheless, one cannot exclude the possibility that the disease gene may not reside at the MRCS locus, even though no positive linkage was found at any of the known microphthalmia loci. Two point linkage and haplotype analysis to adjacent markers refined the putative disease interval (MRCS) to a map distance of 5.0 cM between markers D11S4191 and D11S1883 (Figs 1 and 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic diagram of chromosome 11 indicating the provisional position of the MRCS in relation to the NNO1 locus. The microsatellite markers shown are those that form the haplotypes in Figure 1. The horizontal arrows indicate the boundaries of the critical genetic interval for MRCS on 11q13 as defined by recombination events.
The putative MRCS locus on 11q13 is encompassed by the nanophthalmia locus, NNO1. The phenotypic similarities between this condition and the anomalies segregating in our family suggest allelic heterogeneity and if this were the case, the data would refine the NNO1 locus (originally localised to 14.7 cM region between 11p12 and 11q13) to a 5.0 cM interval on chromosome 11q13 (Fig 4). Within the region lie several genes that are expressed in the eye, ROM1, VMD2, GNG3, FLJ20539, and B3GAT3. ROM1 encodes a protein found in the outer segment disc membranes of the photoreceptor cells of the retina and, together with peripherin/RDS gene, has been implicated in the pathogenesis of digenic retinitis pigmentosa.35 Although ROM1 has not been shown to have a developmental role, we felt it warranted exclusion as a candidate gene. Direct sequencing of the coding and promoter regions of the gene however did not identify a mutation.
The family reported in this paper has a rare ocular phenotype consisting of microcornea, rod-cone dystrophy, cataract, and posterior staphyloma (MRCS). This is likely to be caused by a mutation in a major developmental regulator gene and molecular genetic analysis suggests that the gene locus may lie within the nanophthalmos locus on chromosome 11, possibly allelic with the as yet uncharacterised NNO1 gene. We now plan to try and identify additional families with this rare syndrome, which may help confirm the locus, and screen other candidate genes within the genetic interval. Identification of the causative gene is likely to contribute significantly to understanding the mechanisms underlying human eye development.