Article Text

Abstract
Background: Besides the three known genes (RHO, RDS/Peripherin, NRL) involved in autosomal dominant retinitis pigmentosa (adRP), a fourth gene, RP1, has been recently identified. Initial reports suggest that mutations in the RP1 gene are the second most frequent cause of adRP. The clinical findings were described in a family with adRP and a novel mutation in the RP1 gene.
Method: Index patients from 15 independent families with adRP in which RHO mutations had been excluded in previous examinations were screened for mutations in the RP1 gene by means of direct DNA sequencing. Evaluation of the RP1 phenotype in patients included funduscopy, kinetic perimetry, dark adapted final threshold test, standard electroretinography and, in one case, multifocal electroretinography.
Results: One novel nonsense mutation (Lys778ter) in one of these 15 patients was detected. Cosegregation of the mutation with the disease phenotype could be established in the index patient's family. The phenotype comprises variable expression of clinical disease probably including one case of incomplete penetrance, a onset of symptoms beginning in adulthood, and evidence of regionally varying retinal function loss.
Conclusion: The Lys778ter mutation localises inside the critical region harbouring all mutations described so far. The ophthalmic findings support previous observations that variation of disease expression appears as a typical feature of the RP1 phenotype.
- RP1 gene
- mutation
- autosomal dominant retinitis pigmentosa
Statistics from Altmetric.com
Retinitis pigmentosa (RP) is a clinically and genetically heterogeneous group of inherited retinal degenerations with an estimated worldwide prevalence of approximately 1 in 3500 individuals.1 The term RP refers to the typical ophthalmoscopic finding of pigmentary, bone spicule-like deposits in the mid-peripheral fundus. The course of the disease is characterised by night blindness followed by progressive constriction of the peripheral visual field and, later, loss of central vision leading eventually to blindness in many cases. Typically, rod photoreceptors are primarily affected, evidenced by a severely abnormal or non-detectable scotopic electroretinogram (ERG) even in early stage disease, whereas cone receptor function is secondarily compromised as the disease proceeds.1,2
RP can be inherited in an autosomal dominant, autosomal recessive, X linked, and digenic mode and several genes implicated in these differently inherited forms of adRP have already been identified.3,4 In addition to the (three) known genes (RHO, RDS/Peripherin, and NRL5–8) a fourth gene, RP1, involved in autosomal dominant RP was recently identified.9–11 Based on genetic linkage analysis in the two large families, UCLA-RP01 and Australian D, the RP1 locus has been initially mapped to a 4 cM interval on chromosome 8q11–13,12–14 and the gene has been identified by positional candidate gene analysis. The RP1 gene consists of four exons, gives rise to a transcript of approximately 7 kb, and encodes a protein of 2156 amino acids. The function of the RP1 protein is still unknown even though some possible roles have been suggested. A region of the RP1 protein bears some homology to the human doublecortin (DCX) protein15,16 and to the Drosophila melanogaster protein bifocal.17 These homologies might suggest a role in photoreceptor metabolism11 and/or development and differentiation of the retina.10,18 The presence of a leucine zipper and three nuclear localisation motifs indicate that the protein exerts its function within the nucleus.10,19 Recent screening data suggest that mutations in the RP1 gene are responsible for at least 7%20 of adRP cases in general and for approximately 4% of adRP in the United States and Canada.11
Several mutations in the RP1 gene have been identified,9–11,20,21 many of them clustering in a small region of the fourth exon.20,22 Most RP1 mutations cause a premature termination resulting in severely truncated proteins.9–11,23
In this study we report the screening of the RP1 gene in 15 independent adRP patients enrolled at the University Eye Hospital Tübingen. We identified a novel heterozygous nonsense mutation in the RP1 gene localised inside the common mutation cluster20 and investigated its clinical phenotype.
PATIENTS AND METHODS
Recruitment of patients, DNA isolation, and mutation analysis
The entire coding region (exons 2, 3, and 4) of the RP1 gene was sequenced in 13 German patients from independent families, one Spanish patient, and one patient originating from Italy, all suffering from autosomal dominant retinitis pigmentosa, in which mutations in the rhodopsin gene had been excluded in previous screenings.
All patients had been diagnosed at the Universitäts-Augenklinik Tübingen on the basis of typical symptoms and signs—that is, bilateral progressive disease, night blindness and peripheral visual field loss, fundus findings of optic disc pallor, attenuated retinal vessel and peripheral pigmentary changes, and a severely abnormal or extinguished rod ERG.
Inheritance was considered autosomal dominant when the disease phenotype was observed in at least two subsequent generations.
DNA was isolated from peripheral venous blood samples using the procedure of Miller et al.24 Coding exons 2, 3, 4 and flanking intronic/untranslated regions were amplified from total genomic DNA by means of the polymerase chain reaction (PCR). The sequences of the oligonucleotide primer pairs were derived from the published gene sequence (GenBank accession number AF128525). Exons 2 and 3 were amplified as single fragments with primer pairs 37486F/38320R and 38601F/39015R, respectively, using the AmpliTaq polymerase (Perkin Elmer). Exon 4 was also amplified as one single fragment (primer pairs 41118F/47616R) but using the expand long template PCR system (Boehringer Mannheim, Germany). All PCR reactions were performed under appropriate buffer and cycling conditions.
Amplified PCR products were purified by ultrafiltration using Centricon-100 columns (Millipore) according to protocols recommended by the manufacturer.
Exons 2, 3, and 4 were bidirectionally sequenced employing the big dye terminator chemistry (PE Biosystems). Sequences were all analysed with an automated ABI 377 DNA sequencer (Applied Biosystems, Foster City, CA, USA) using the fluorescent dideoxynucleotide technology. For sequence editing and alignments the lasergene software package (Dnastar, Madison, WI, USA) was used.
Cosegregation analysis was performed by sequencing a PCR amplified subfragment of exon 4. Sequences of oligonucleotides used for PCR reactions and DNA sequencing can be obtained from the authors on request.
Clinical studies of the RP1 mutation phenotype
Outside medical records were obtained from two family members carrying and from one person not carrying the RP1 nonsense mutation; three affected family members were clinically investigated for phenotype after informed consent had been obtained. Phenotype analysis comprised clinical routine examination, kinetic perimetry, measurement of dark adaptation final thresholds, Ganzfeld electroretinography and, in one individual, multifocal electroretinography. Kinetic visual fields were recorded with targets V/4e, III/4e, and I/4e of a Goldmann perimeter. Dark adapted final thresholds were measured after a period of 45 minutes of dark adaptation with blue-green (500 nm) and red (656 nm) targets (2 degree in diameter) of a Tübingen perimeter. Thresholds were determined with an ascending method of limits by increasing target luminance in 1 dB steps.25 Scotopic and photopic Ganzfeld electroretinography was recorded according to the ISCEV standard 26 with an UTAS 2000 system (LKC Technologies, Gaithersburg, USA), and DTL electrodes. White flashes were used, the intensity of the standard flash was 1.7 cds/m2. Multifocal ERG (mfERG)27 was performed according to a previously published protocol28 with the veris 3.1 system (EDI, San Francisco, CA, USA). Briefly, mfERG responses were elicited by an array of 63 black or white hexagonal elements scaled in size by eccentricity, entirely covering a visual field of approximately 30 degrees. The stimulus was presented at a viewing distance of 28 cm on a monitor with a frame rate of 75 Hz, a mean luminance of 51.8 cd/m2 and a black/white contrast of 93%. Amplitudes and peak times of the first positive deflection (P1) were determined for the foveal element and as the averaged responses of four peripheral regions grouped in concentric rings around the foveal element.
RESULTS
DNA analysis
RP1 mutations were ruled out in 14 of the 15 patients under analysis, one patient (Fig 1B, IV:1) showed a heterozygous A to T transition in exon 4 resulting in a novel nonsense mutation Lys778ter (AAA→TAA) (Fig 1A). This mutant allele is predicted to encode a severely shortened RP1 protein of 777 amino acids, 1379 amino acids less than the wild type RP1 protein.
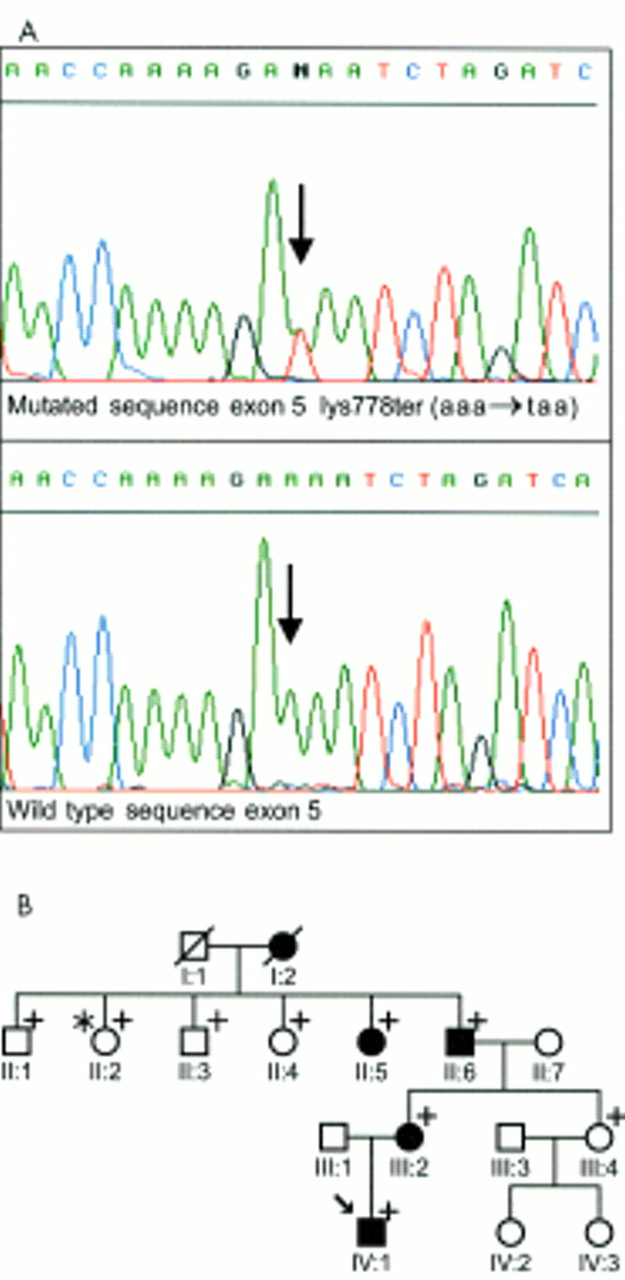
(A) Detection of the novel Lys778ter mutation in the RP1 gene by sequence analysis. The electropherogram representing the mutated sequence is shown on top while the wild type sequence is shown below. The arrow indicates the heterozygous Lys778ter mutation. (B) Pedigree with the Lys778ter mutation in the RP1 gene. Solid symbols indicate family members with adRP while open symbols represent healthy individuals. Subjects for whom samples were available for molecular analysis are indicated by a plus sign; one individual (II:2) with apparent incomplete penetrance is marked with an asterisk; the index patient (IV:1) is additionally marked with an arrow.
Analysis of codon 778 in four (II:5; II:6; III:2; IV:1) affected and four (II:1; II:3; II:4; III:4) (Fig 1B) unaffected family members revealed that all affected members were heterozygous for this mutation, confirming the cosegregation of the Lys778ter mutation with the disease phenotype in this family. However, the mutation was also detected in one apparently healthy family member (II-2).
In addition to this novel mutation we detected six polymorphisms, of which all except the first one had already been described.22 They are as follows: Arg872His (CGT→CAT) (31%), Asn985Tyr (AAT→TAT) (56%), Ala1670Thr (GCA→ACA) (19%), Ser1691Pro (TCT→CCT) (25%), Gln1725Gln (CAA→CAG) (31%) and Cys2033Tyr (TGT→TAT) (56%).
Phenotype of the Lys778ter mutation
Figure 1B shows the pedigree of the family investigated. Four individuals not carrying the mutation (II:1; II:3; II:4; III:4, age range 48–67) do not show signs of RP on funduscopy, Goldmann perimetry (all) or scotopic, and photopic flash ERG (performed in II:4 only), according to external medical records.
Individual I:2 was reported to disclose typical symptoms of retinitis pigmentosa from the end of her fifth decade of life. Medical records were not available. Case II:2, a 60 year old female, carries the mutation, but has no visual complaints, nor, according to outside medical records, does she show clinical signs of RP—that is, her fundus appears normal and she has normal visual fields and visual acuity. Case II:5, a 68 year old female, likewise is unaware of typical symptoms related to RP. However, external records reveal that, on funduscopy, she has peripheral pigmentary degeneration characteristic of RP, and that kinetic visual fields (Goldmann target III/4e) of both eyes are constricted to about 20–30 degrees. Visual acuity is 20/40 in both eyes. There is nuclear cataract and asteroid hyalosis in both eyes.
ERG records are not available for II:2 or II:5.
Case II:6, a 72 year old male, noticed peripheral vision problems as the presenting symptom at about age 45, followed by night blindness at about age 50. At that time the diagnosis of RP was established. Central visual function—that is, reading acuity, contrast sensitivity, and colour vision have been deteriorating since the last decade, mainly in the left eye.
On examination, visual acuity was 20/30 right eye and light perception left eye. Perimetry revealed only a small central island (right eye) and no comprehensive field left eye. Dark adapted final threshold tested at 6 degrees nasally in the right visual field was elevated by about 2 log units for the 500 nm, and 1 log unit for the 656 nm target. The threshold difference between the blue-green and red target revealed residual rod function at this retinal location. On Ganzfeld ERG both rod and cone signals were within noise level. Biomicroscopy showed small lens opacities in both eyes, and typical funduscopic signs of advanced stage RP affecting all peripheral quadrants.
Case III:2, the daughter of II:6, is a 49 year old woman with a history of night blindness since about age 35, and peripheral field loss since about age 40. In the past 5 years she has experienced a rapid decline of visual acuity in both eyes.
Clinical examination disclosed a visual acuity of 20/400 in both eyes. On Goldmann perimetry there were severely constricted central islands and small field remnants in the outer temporal periphery in both eyes. The dark adapted final threshold tested at a central retinal location (2 degrees nasally right eye) showed an elevation by about 3 log units for 500 nm and 2 log units for 656 nm, and pure cone mediation evidenced by equal 500 and 656 nm thresholds. Rod and cone ERG signals were non-detectable. Biomicroscopy revealed dense asteroid hyalosis right eye and cortical cataract left eye, and fundus findings typical of advanced RP in all retinal quadrants of both eyes.
Case IV:1, the affected 22 year old son of III:1 has a history of esotropia and amblyopia right eye. He reported problems with dim light conditions and a decrease of visual acuity beginning at age 18. Peripheral visual field defects had been detected 1 year ago. Clinical evaluation revealed a visual acuity of 20/400 right eye and 20/70 left eye. Visual field was near normal to the V/4e target in the left eye, but showed depressions in the temporal, mainly the upper temporal, fields to V/4e in right eye, and to the III/4e and I/4e targets in both eyes, respectively (Fig 2). Dark adaptation final threshold, determined at 20 degrees nasally in the left visual field, was clearly rod mediated and showed only a slight elevation (0.6 log units above normal for 500 and 656 nm). On flash ERG (Fig 2), isolated rod b-waves were very small in the right, and not detectable in the left eye, scotopic mixed b-wave responses were clearly discernible from noise but markedly reduced (about 20% of the median in right eye and 10% in left eye, respectively).

Phenotypic features of patient IV-1 carrying the Lys778ter mutation. Top: kinetic visual fields. Bottom left: standard ERG curves (right lane), compared to those of a normal observer (left lane). Wave forms, from top to bottom, are: scotopic isolated rod b-wave, scotopic mixed rod/cone response, photopic single flash cone response, photopic 30 Hz flicker response. Arrows indicate the peaks of b-waves or the 30 Hz flicker signals, respectively. Bottom right: fundus of the left eye showing the nasal periphery (upper photograph) and the posterior pole.
Photopic cone signals (b-waves and 30 Hz flicker signals) were slightly better preserved than scotopic b-waves (about 25% of amplitude median right eye, and 20% left eye), their peak times were markedly delayed.
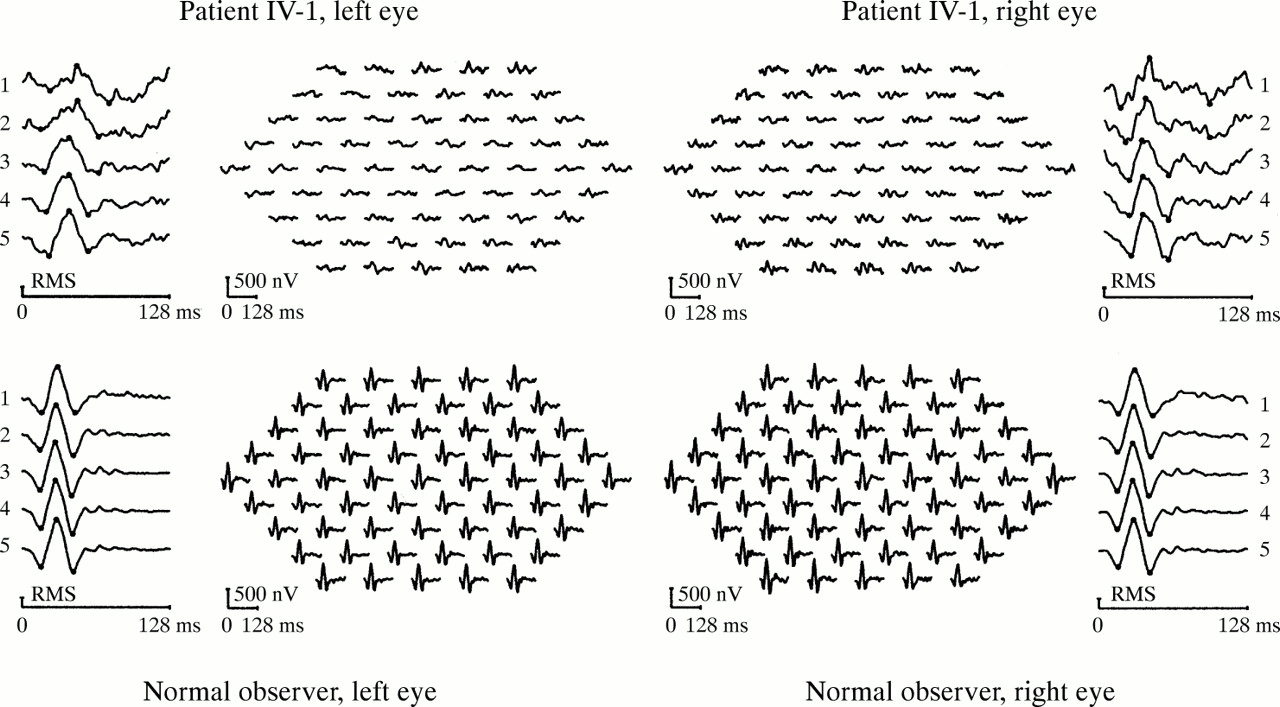
Multifocal ERG signals (Fig 3) were reduced all over the test field in both eyes. The topography of abnormalities paralleled the visual field findings in that amplitude losses tended to be more severe in the temporal than in the nasal fields. The smallest amplitudes were found in the macular and perimacular regions (rings 1 and 2), and amplitudes increased towards the periphery of the field. Likewise, the timing was prolonged at all eccentricities; however, the strongest delay was observed in the two most central regions, whereas the timing of the responses shortened with eccentricity, reaching near normal values at the field border of the right eye.

{kind=link}
{kind=link}
{kind=link}
Multifocal ERG of patient IV:1 (top), compared to a normal observer (bottom). The outer panels show averaged signals grouped concentrically by eccentricity (No 5 being the most peripheral group). The central panels show the trace arrays composed of 61 local retinal responses. A predilection of function loss in the macular and the temporal pericentral areas can be seen.
Biomicroscopy of anterior segments was unremarkable. Funduscopy (Fig 2) showed cystoid macular oedema in both eyes. Peripherally, vessel attenuation, RPE atrophy, and bone spicules were prominent in the nasal fundus, whereas the temporal fundi showed only slight RPE changes and barely any pigmentation.
DISCUSSION
We have analysed patients from 15 independent families showing typical clinical features of autosomal dominant RP. In previous studies all of them had been screened for mutations in the rhodopsin gene and found to be negative. Even though mutations in the most affected gene in adRP, rhodopsin, have been excluded, we could detect only one mutation in these patients, mainly originating from Germany, when screening the entire coding region of the RP1 gene. This fact leads to the assumption that the frequency of mutations in the RP1 gene in German patients might be relatively small. The current sample size; however, is too low to draw more exact conclusions about the proportion of adRP cases that are caused by RP1 mutations.
The RP1 gene is a very long gene consisting of approximately 7 kb. Because of its enormous size and the low frequency of mutations the ratio between the effort of screening the entire gene and the probability to find a mutation seems to be quite small.
A different study20 reports that mutations in the RP1 gene are clustered in a small region in exon 4 spanning codon 650 to 796. The novel Lys778ter mutation localises inside this mutation cluster region. But recently other studies have found mutations outside this critical segment and have therefore proposed to extend the region of mutational screening.22,23 In conclusion we suggest to analyse an interval spanning from codons 500 to 1053.
The phenotype in the Lys778ter family shows a few features that deserve to be mentioned and may help to define common characteristics of RP1 mutation phenotypes.
First of all, there is considerable variation of disease expression, as to subjective onset of symptoms (range: age 18 up to “asymptomatic” at age 68) and clinical disease. Fundus findings and peripheral visual field loss typical of RP were observed at the earliest in 22 year old patient IV:1. The other extreme is 60 year old patient II:2, who on the basis of the clinical findings available might be classified as a case of non-penetrance. It cannot be excluded, however, that this latter patient would have shown minimal disease expression on ERG testing. Jacobson et al22 found slight reductions in rod ERG maximum amplitude in clinically otherwise normal heterozygote carriers of the RP1 Arg677ter mutation. Variation of disease expression, including cases of non-penetrance, has been reported before among RP1 families10,20 and could also be observed in a larger sample of patients carrying the same Arg677ter mutation,22 thus emerging as a typical feature of RP1 associated RP.
Despite the variability of disease severity displayed among carriers of the Lys778ter mutation attempts can be made to describe general features of the phenotype. Onset of symptoms did not occur before adulthood. Night blindness, if the presenting symptom, did precede visual field problems by only a few years. In comparison, a number of adRP phenotypes caused by rhodopsin mutations have earlier, childhood, onset of night blindness.29 Some families with Arg677ter, or a 2280–2284 del mutation of RP1 have likewise been classified as “late onset” phenotypes.20
Autosomal dominant forms of RP may clinically differ as to the retinal topography of disease and the relation of rod to cone function loss.29–31 Although the data are limited, some conclusions may be drawn from the clinical, psychophysical, and ERG findings in the Lys778ter family. In two older individuals (II:6 and III:2), the common unspecific features of advanced stage disease appear funduscopically, by concentric visual field loss, and by flat ERGs. In an earlier disease stage, however, (IV:1) a regional predelection of retinal damage is revealed. Peripheral fundus changes are visible mainly in the nasal regions, and both multifocal ERG and perimetry show losses in the corresponding temporal or the upper temporal field, respectively. Based on the standard ERG findings, the rod system overall appears to be substantially affected, and more affected than the cone system. Psychophysical rod final thresholds, however, give evidence of near normal sensitivity at least in single retinal locations. Interestingly, rod function is psychophysically detectable even in late stage disease (72 year old individual II:6) with small residual field islands. These findings argue rather for a regionally evolving pattern of rod disease in RP1 Lys778ter than a primary diffuse rod defect.
Our impressions are paralleled by the detailed investigations of Jacobson et al22 on a group of patients carrying the Arg677ter or various RP1 frameshift mutations. They describe a common pattern, comprising regional retinal variation of disease with predilection of the inferonasal part, both for the rod and the cone system, and with rod function being more affected than cone function at all disease stages.
Moreover, they concluded from the cross sectional data and from individual follow up measurements, that progression of rod function loss over time is faster than that of cone function loss. They emphasised good preservation of central cone function even until very late disease stages in almost all patients.
The latter findings differ in our family. The majority of patients, including the youngest, have partly severe visual acuity loss in one or both eyes. In IV:1, central cone function was studied in more detail. The multifocal ERG showed an atypic pattern. The centroperipheral gradient of amplitudes and peak times normally observed in RP (that is, signals become smaller and slower with eccentricity)32 was inverted. The abnormalities became more prominent towards the fovea, clearly indicating severe affection of macular cone function. It remains uncertain, however, to what extend the central function loss in this case is due to the cystoid macular oedema, an associated condition not specific for a single RP genotype.
Further clinical studies are necessary to study the influence of cystoid macular oedema on the multifocal ERG of RP patients and to evaluate the risk of macular function loss in RP1.
By now, prognostic counselling in patients carrying RP1 mutations should take into account a high variability in severity of both peripheral and macular disease processes.
Acknowledgments
We thank all patients and family members for participating in this study. We also thank Dr Carl Erb for providing fundus photographs and Ms Katrin Vohrer for technical assistance.
This work was supported by the Edeltraud-Blickle-Stiftung and by a grant from the Deutsche Forschungsgemeinschaft (SFB 430/ C2) to EA-S and EZ.