Article Text

Abstract
AIMS To compare the clinical and molecular genetic features of two phenotypically distinct subgroups of families with type 1 Stickler syndrome.
BACKGROUND Stickler syndrome (hereditary arthro-ophthalmopathy, McKusick Nos 108300 and 184840) is a dominantly inherited disorder of collagen connective tissue, resulting in an abnormal vitreous, myopia, and a variable degree of orofacial abnormality, deafness, and arthropathy. Stickler syndrome is the commonest inherited cause of rhegmatogenous retinal detachment in childhood with a risk of giant retinal tear (GRT) which is commonly bilateral and a frequent cause of blindness.
METHOD Pedigrees were identified from the vitreoretinal service database and subclassified according to vitreoretinal phenotype. Ophthalmic, skeletal, auditory, and orofacial features were assessed. Linkage analysis was carried out with markers for the candidate genes COL2A1, COL11A1, and COL11A2. The COL2A1 gene was amplified as five overlapping PCR products. Direct sequencing of individual exons identified mutations.
RESULTS Eight families exhibiting the type 1 vitreous phenotype were studied. Seven were consistent for linkage to COL2A1, with lod scores ranging from 2.1 to 0.3. In most instances linkage to COL11A1 and COL11A2 could be excluded. One family was analysed without prior linkage analysis. Three of the families exhibited a predominantly ocular phenotype with minimal or absent systemic involvement and were found to have mutations in exon 2 of COL2A1. Five other pedigrees with an identical ocular phenotype plus orofacial, auditory, and articular involvement had mutations in others regions of the COL2A1 gene. None of the pedigrees exhibited the characteristic lenticular, retinal pigment epithelial, or choroidal changes seen in Wagner syndrome.
CONCLUSIONS These data confirm that type 1 Stickler syndrome is caused by mutations in the gene encoding type II collagen (COL2A1). In addition, data are submitted showing that mutations involving exon 2 of COL2A1 are characterised by a predominantly ocular variant of this disorder, consistent with the major form of type II procollagen in non-ocular tissues having exon 2 spliced out. Such patients are all at high risk of retinal detachment. This has important implications for counselling patients with regard to the development of systemic complications. It also emphasises the importance and reliability of the ophthalmic examination in the differential diagnosis of this predominantly ocular form of Stickler syndrome from Wagner's vitreoretinopathy.
- COL2A1
- Stickler syndrome
- vitreous
- Wagner syndrome
Statistics from Altmetric.com
Stickler syndrome (hereditary arthro-ophthalmopathy, McKusick Nos 108300 and 184840) is a dominantly inherited disorder of collagen connective tissue,1 2 resulting in an abnormal vitreous and a variable degree of orofacial abnormality, deafness, and arthropathy. It is the commonest inherited cause of rhegmatogenous retinal detachment in childhood. Although the systemic features are widespread, the sight threatening complications are perhaps the most conspicuous and serious manifestations, particularly the risk of giant retinal tear (GRT) which is frequently bilateral and, if untreated, leads to blindness.
Abnormalities of vitreous gel formation and architecture are pathognomonic of Stickler syndrome3 and two distinct vitreoretinal phenotypes are now recognised. The majority of families show a characteristic congenital “membranous” vitreous anomaly (type 1) but in a minority a different “beaded” (type 2) phenotype is apparent.4-7 All families with full Stickler syndrome exhibit an abnormal vitreoretinal phenotype, but other systemic changes can be highly variable both between and within families.
The first mutation in a family with Stickler syndrome was identified in COL2A1,8 the gene for type II collagen, a major component of both cartilage and vitreous. The original kindred reported by Stickler also had a mutation in this gene.9 Both resulted in premature termination codons which appear to be a distinctive feature of the Stickler phenotype compared with other inherited connective tissue dysplasias associated with COL2A1 mutations.10 It is now known that the type 1 vitreous phenotype, as described by Scott,4 correlates with defects in type II procollagen (Snead et al 5 and unpublished data).
Pedigrees with other varieties of vitreous phenotype are genetically heterogeneous. Some have mutations in COL11A1 the gene for α1(XI) collagen, one of two type XI α chains, which form heterotypic fibrils with type II collagen.6 7 These pedigrees feature sparse and irregularly thickened bundles of “beaded” fibres (type 2 vitreous phenotype) throughout the vitreous cavity. It is not yet clear whether this is specific to mutations in COL11A1 or may also result from defects at other loci. The other polypeptide chain (α2) of type XI collagen is not expressed in the vitreous11 so that mutations in its encoding gene (COL11A2) produce a syndrome with the systemic features of Stickler syndrome but without eye involvement.12-16 Evidence for another Stickler syndrome locus also exists.7 17
Type II procollagen exists in two alternatively spliced forms. A short form, which is expressed in cartilage, has exon 2 spliced out, resulting in a molecule with a smaller N-propeptide (αIIb). The short form also functions as a third α(XI) chain in cartilage. It co-assembles with products of the COL11A1 and COL11A2 genes, to form a α1(XI) α2(XI) α1(IIb) heterotrimer. Thus, mutations in COL2A1 can affect both type II and XI collagen in this tissue.18 In vitreous both transcripts exist but here the longer form predominates.19 It is unclear whether this longer form can also participate in heterotrimer formation with α1(XI) collagen
Of the Stickler syndrome pedigrees exhibiting the type 1 vitreous phenotype, a subgroup has also been identified that is characterised by a predominantly ocular disorder without systemic skeletal or auditory involvement. Before the introduction of the subclassification of Stickler syndrome based on vitreous phenotype it is possible that some of these pedigrees might otherwise have been confused with another inherited vitreoretinopathy, Wagner syndrome (MIM 14320020 21). However, although Wagner syndrome also appears to be a predominantly ocular vitreoretinopathy without systemic manifestations, it has distinct clinical differences from Stickler syndrome,22 23 and in some families linked to a separate locus on chromosome 5.22 24 25 The term “Wagner-Stickler” syndrome,26-29 is therefore confusing and should be abandoned.
Here we describe the clinical and molecular genetic features of eight families with Stickler syndrome. All eight families feature the same type 1 vitreous phenotype but three of the families have a predominantly ocular phenotype with minimal or absent systemic involvement and were found to have mutations in exon 2 of COL2A1. Five other pedigrees have, in addition to the type 1 ocular phenotype, auditory and articular involvement and have mutations in others regions of the same gene. None of the pedigrees exhibits the characteristic lenticular, retinal pigment epithelial, or choroidal changes seen in Wagner syndrome.22 23
Method
CLINICAL EXAMINATION
Pedigrees were identified from the vitreous clinic database at Addenbrooke's Hospital and subclassified prospectively according to vitreoretinal phenotype before molecular genetic analysis. Informed written consent was received in all cases and prior ethical approval for the study was obtained (LRC92/019).
OPHTHALMIC EXAMINATION
A general ophthalmic history was recorded with particular attention to the age of onset, degree and progression of myopia, cataract, and vitreoretinal disease. A full ophthalmic examination including refraction, slit lamp biomicroscopy, indirect ophthalmoscopy, and B scan ultrasonography was carried out. In some of the younger patients applanation tonometry and gonioscopy were not possible. Anterior and posterior segment photographs were taken where appropriate.
PHYSICAL DEVELOPMENT
In order to assess the developmental morphology of the face and hands, all patients were examined subjectively and objectively. A subjective assessment was made of the non-quantifiable factors retrognathia and anteversion of nares. All other factors were measured objectively according to a standardised method.30Anteroposterior and lateral facial photographs were taken at a standardised scale of 1:8, and of both hands at a scale of 1:10 using a Nikon FM2 camera with Micro Nikon 105 mm medical lens and Kodachrome 64 film at F16. A 1 cm grid was printed and then photographed at a scale of 1:8 to match the facial films and 1:10 to match the hand views. Clinical measurements of outer canthal distance, inner canthal distance, philtrum length, and middle finger length were also recorded at the time of initial examination. These were used subsequently to test the accuracy of the method of photographic calibration. Control measurements of inner and outer canthal distance, interpupillary distance, and philtrum length from 20 unaffected siblings and 60 age matched controls (recruited from the general ophthalmic clinic) were also recorded.
Using dual projection of identically calibrated projectors, the matched scales were superimposed over each facial view in turn so that the quantifiable facial measurements could be made. This was repeated using the matched scale for the hand views. All measurements were made according to published criteria.30
GENERALISED SKELETAL COMPLEX
Height and weight were measured and an assessment made of general body build. Joint hypermobility was assessed objectively using the Beighton scoring system. A score of one or zero is given for a series of joint manoeuvres and the total sum allocated up to a possible maximum score of 9/9. This method allows comparison with published age and sex matched populations.31 Plain anteroposterior and lateral radiographs were carried out on affected pedigree members.
AUDIOLOGY EXAMINATION
A specific inquiry was made regarding the date and progression of any subjective hearing loss and in particular whether this had been a congenital, sudden, or progressive deterioration. A record of the type and duration of noise exposure was made together with any contributory factors such as glue ear, grommets, or drug induced sensorineural damage. All affected patients underwent bilateral otoscopy and audiometry involving air and bone conduction testing according to standardised procedures.32
MOLECULAR GENETIC ANALYSIS
Seven families with the type 1 vitreous phenotype were used for linkage analysis to markers for COL2A1, COL11A1, and COL11A2 as previously described.7 All were consistent for linkage to COL2A1, with lod scores ranging from 2.1 to 0.3. In most instances COL11A1 and COL11A2 could be excluded (data not shown). Another family with the type 1 vitreous phenotype was used without any linkage analysis. All eight were analysed for COL2A1 mutations.
COL2A1 AMPLIFICATION AND SEQUENCING
Database available DNA sequences were used to design primers for polymerase chain reaction (PCR) amplification. A 30.991 kb sequence contained exons 1–54 (Accession No L10347); 343 bp of 3′ untranslated region was contained in an EST Image clone (Accession No H62509). Five PCR products covering bases (1) 4052–6902, (2) 6028–11591, (3) 11331–19598, (4) 18342–26231, (5) 24171–30991+106 3′UTR were amplified using DNA from a second or third generation affected individual. In each case the primers for PCR corresponded to the first 24 sense and last 24 antisense nucleotides of each product. Amplification reactions used TaqPlus (Stratagene) as previously described.7 Products were purified using QIAquickspin columns (Qiagen), and eluted in water. Direct cycle sequencing of individual exons contained within the PCR products was performed with BigDye terminator RR mix (PE Applied Biosystems). Sequencing primers were situated around 50 bp up and downstream from each exon and were derived from the published sequence for the COL2A1 gene (Accession NoL10347). Reactions were analysed using an ABI 377 machine.
FAMILY DNA ANALYSIS
Smaller regions of COL2A1 containing potential mutation sites, identified by sequencing, were amplified, from each family member. Taq DNA polymerase (Life Technologies) was used with amplification conditions described previously.6 Products were purified as above and used in a 33P labelled single dideoxynucleotide sequencing reaction, such that the base change under test could be detected. Thermosequenase version II (Amersham International) was used for the direct sequencing reaction. Products were separated by electrophoresis in a denaturing polyacrylamide gel and then autoradiographed. Spouses marrying into the family were used as normal controls.
Results
The type 1 vitreous anomaly was present in all affected individuals and clinical status was confirmed on subsequent mutation analysis (see below). Although there was both interfamilial and intrafamilial variation in systemic phenotype the vitreous phenotype remained consistent throughout and this emphasises the importance of vitreous slit lamp examination in this disorder. Five families (MS2, MS18, MS20, MS54, and MS66) expressed significant auditory and skeletal abnormalities in addition to their ocular changes. The remaining three families (MS11, MS13, and MS62) expressed a predominantly ocular phenotype in which skeletal, auditory changes were minimal and orofacial abnormalities were present only in a minority of individuals. These differences are illustrated and quantified in Tables 1 and 2. In seven families linkage analysis was consistent with COL2A1 as the disease locus. An additional family (MS54) was analysed solely on the basis of the vitreous phenotype.
Phenotypes resulting from COL2A1 mutations not in exon 2. Clinical manifestations were scored as 0 absent, 1 mild, 2 moderate, and 3 severe, except for retinal detachments which was scored 1 single eye or 2 both eyes
Phenotypes resulting from COL2A1 exon 2 mutations. Clinical manifestations were scored as 0 absent, 1 mild, 2 moderate, and 3 severe, except for retinal detachment, which was scored 1 single eye or 2 both eyes
The COL2A1 gene was amplified as five overlapping PCR products and individual exons were directly sequenced. In all cases changes to the sequence which lead to premature termination codons and haploinsufficiency were found (examples are shown in Fig 1). Five of these were frame shift mutations and three were single base substitutions (Table 3). Mutations in each family were confirmed by single dideoxynucleotide sequencing reactions designed to detect the abnormality. In each case the status of affected and normal individuals, as determined by slit lamp examination before molecular analysis, was confirmed by mutation detection. All but one of these mutations are novel and, because they result in premature termination codons, they would be very unlikely to occur in the general population.

Mutation detection by sequencing. Mutations in exon 2 are shown for families MS11, MS13, and MS62 along with the normal sequence (N). The sequence for each is written under the chromatograms. For the frameshifts the mutant sequence is shown from the point at which the sequence becomes heterozygous, is in bold type, and is also indicated in the normal sequence. A 2 bp deletion and 4 bp duplication are boxed. The translated products are shown above and below the normal and mutant sequences respectively. The premature stop codons resulting from the frameshifts are underlined.
Type and position of COL2A1 mutations. The position and type of mutations found in COL2A1 are indicated by nucleotide location within the COL2A1 gene (Accession NoL10347) Amino acids are numbered by the usual convention in which the first glycine of the triple helical region is 1. Amino acids in the N-propeptide are numbered from the initiating methionine
Three mutations were in exon 2. Two of these were frame shift mutations, a 2 bp deletion, and a 4 bp duplication (GGAT) (Fig 2). Both lead to a premature termination codon at the same position in exon 2. The third exon 2 mutation was a single base substitution, which converted a codon for tryptophan to a premature termination codon. Two other frame shifts were both in exon 42 and were insertions of a single cytosine residue into different poly C sequences. These lead to premature termination codons in different positions in the cDNA sequence. Another frameshift mutation was caused by a 1 bp deletion in exon 21. This led to a downstream premature termination codon, in exon 28. Two single base substitutions converting codons for arginine to stop codons were seen in exons 35 and 42. The stop codon mutation in exon 42 was identical to the first Stickler mutation characterised by Ahmad et al in 1991.8

Family confirmation of frameshift mutations.33P labelled single dideoxynucleotide, sequencing reactions (as indicated) were performed on PCR products amplified from individual family members. Numbers indicate nucleotide sequence (Accession NoL10347). Asterisks indicate extra bands seen in affected family members.

Family confirmation of premature termination codons.33P labelled single dideoxynucleotide, sequencing reactions (as indicated) were performed on PCR products amplified from individual family members. Numbers indicate nucleotide sequence (Accession NoL10347). The extra bands seen in affected members are arrowed.
The three families with exon 2 mutations corresponded to those with minimal systemic changes (MS11, MS13, and MS62) (Table 2). Articular changes were absent in all except an 80 year old member of family MS62. It is of interest and importance to note, however, that the risk of ophthalmic complications (myopia, retinal detachment) is high in both groups of families. Although midline clefting was less common in the families with exon 2 mutations the number of patients is too small for statistical analysis.
Discussion
The Stickler and Wagner syndromes are both dominantly inherited vitreoretinopathies and one of the main criteria often used to distinguish between the two is the lack of systemic features in the Wagner syndrome. Wagner20 described a new ocular disease in a three generation pedigree from the Kanton of Zurich with 13 affected individuals. It featured autosomal dominant inheritance, low myopia (3.00 dioptres or less), fluid vitreous, cortical cataract, and inconstant and variably affected dark adaptation. No affected individual suffered a retinal detachment. In a follow up study of Wagner's original pedigree,21 10 further affected individuals were identified. The cardinal features noted were the complete absence of the normal vitreal scaffolding and preretinal, equatorial, and avascular greyish-white membranes. Clear lenses in childhood developed anterior and posterior cortical opacities in puberty and cataracta complicata during the fourth decade. Rhegmatogenous retinal detachment was not reported in either publication but has since been noted in a minority of individuals.33
The association of retinal detachment and cleft palate was noted before Stickler's description of hereditary arthro-ophthalmopathy34 and controversy arose regarding the authenticity of the proposed new syndrome. Indeed, many subsequent publications referred to the “Wagner-Stickler” syndrome,26-29 35 or “Wagner syndrome with arthropathy”36 implying phenotypic variation of the same basic underlying disorder. It has subsequently been shown that families with Stickler syndrome and a non-ocular variant have mutations in the genes COL2A1, COL11A1, and COL11A2; whereas, several families with Wagner syndrome (WGN1) including the original family have been linked to a locus at 5q14.3.24 25
The mutations in COL2A1 exon 2 described here illustrate the critical importance of ophthalmic and particularly vitreous examination in distinguishing between these two disorders, especially if a pedigree is too small to exclude one or other loci on linkage analysis. The main clinical differences are summarised in Table 4.
Comparison of ophthalmic features in type 1 Stickler syndrome and Wagner syndrome
There has been one report of a COL2A1 mutation causing Wagner syndrome.37 However, the clinical description including the mild facial hypoplasia and hypermobile joints more closely resembled that of Stickler syndrome. In our experience the ocular phenotype displayed by COL2A1 exon 2 mutation patients matches that seen in other COL2A1 Stickler syndrome families. Patients exhibit the membranous congenital anomaly and do not show the atrophy of the retinal pigment epithelium and choriocapillaris seen in 5q linked Wagner families (Table 4). In our view these clinical differences are sufficient to distinguish between the two disorders.
Our results also illustrate that the families with COL2A1 exon 2 mutations, even in the absence of obvious skeletal or auditory features are also at high risk of retinal detachment. This is particularly important since the diagnosis of Stickler syndrome may be overlooked or rejected on the basis of the minimal or absent skeletal changes. Although the midline clefting seen in some individuals might suggest Stickler syndrome, most patients have few if any non-ocular characteristics normally associated with this disorder.38
Seven individuals with exon 2 mutations had some degree of midfacial hypoplasia or midline clefting, the latter being severe in two cases. This may indicate that the longer αIIa collagen plays a part during craniofacial development. Certainly, the level of αIIa mRNA is higher in fetal skeletal tissue.18 39 In these few individuals where facial hypoplasia or midline clefting is apparent the diagnosis of Stickler syndrome would be acceptable. However, for the majority of exon 2 mutants (including all members of MS11) the lack of extraocular characteristics might, in the view of some clinicians, exclude the designation of Stickler syndrome. A suggested alternative title might be “predominantly ocular” Stickler syndrome (POSS), commensurate with the non-ocular Stickler syndrome (NOSS) phenotype caused by mutations in COL11A2 and “full” Stickler syndrome (FSS) secondary to other mutations in either COL2A1 or COL11A1.
The absence of arthropathy, cleft palate, hearing loss, and cardiovascular abnormalities led Perveen et al,25 to assign a diagnosis of Wagner syndrome in 18 families. However, 14 were too small to confirm linkage to 5q14.3. The lack of mutations in these families was used to exclude the versican (CSPG2) gene as the Wagner locus. The authors themselves were cautious as to whether all of their families might be linked to 5q14.3. We suggest that unless the retinal phenotype was the same as for families linked to 5q14.3, then COL2A1 should be considered a candidate for the disease locus in families with vitreoretinopathy without systemic changes, unless it could be excluded by linkage. It is not known if mutations in other regions of the COL2A1 gene can also lead to similar ocular only phenotypes.
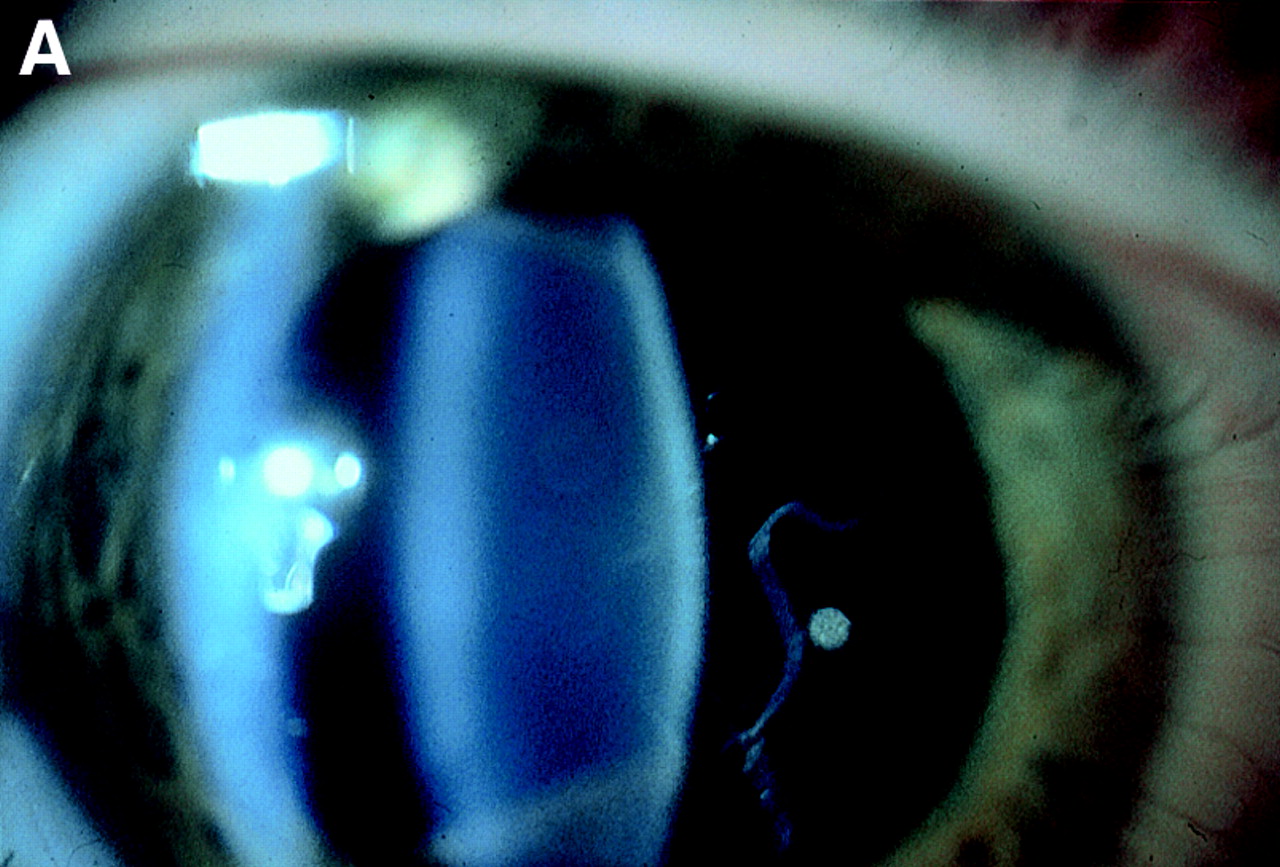
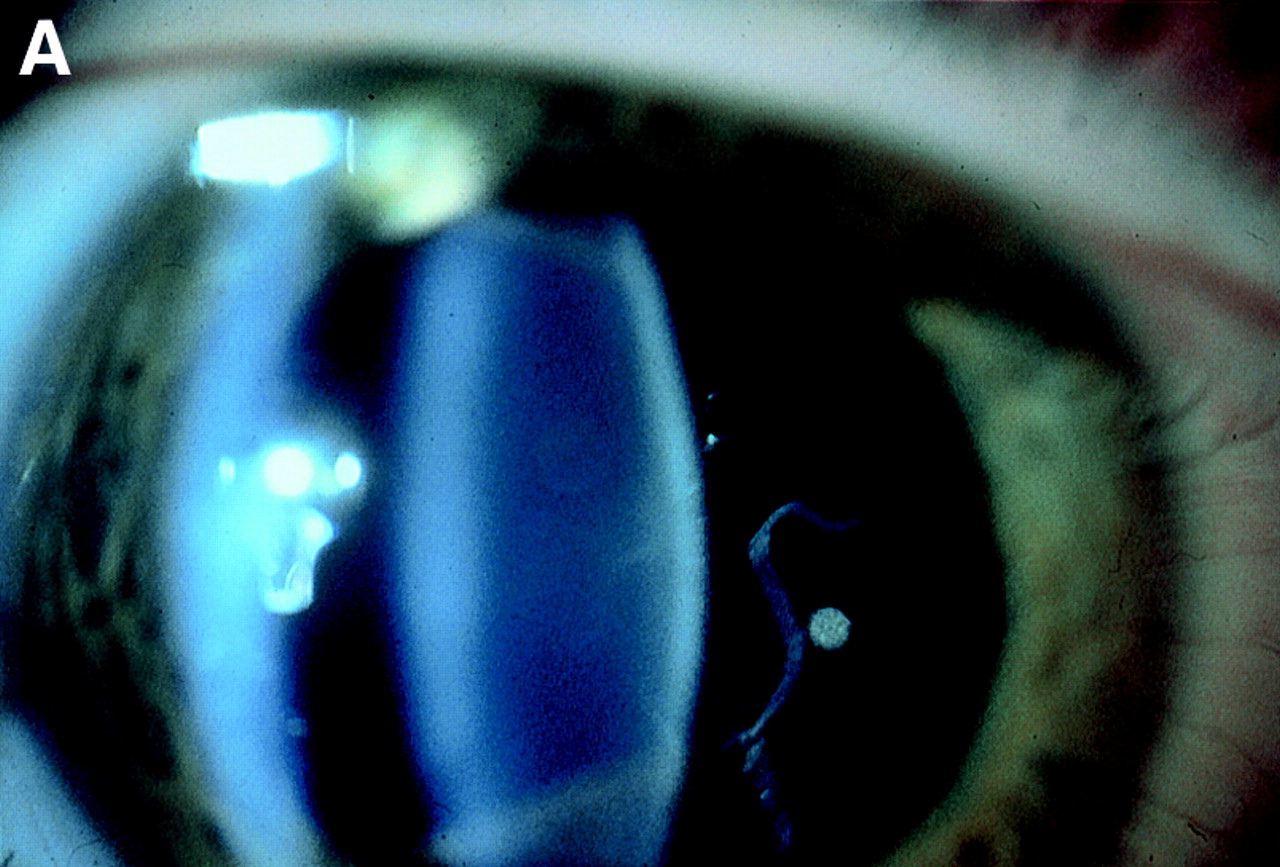
Finally, our family studies emphasise the reliability of vitreous phenotype in the diagnosis of Stickler syndrome. Whereas systemic features vary between and within families the eye changes were remarkably consistent (Fig 4). Of 52 individuals examined (excluding unaffected spouses) mutation detection ratified the clinical diagnosis in 35 affected and 19 unaffected individuals.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Vitreous phenotypes in Stickler syndrome. (A) Type 1 phenotype: COL2A1 non-exon 2 family MS20. (B) Type 1 phenotype: COL2A1 exon 2 family MS13. (C) Type 2 phenotype: COL11A1 mutation (data not shown, family MS42 see Martin et al7).
Acknowledgments
The authors gratefully acknowledge the financial support of The Iris Fund for Prevention of Blindness, The Medical Research Council, The Stanley Thomas Johnson Foundation, and Michael and Elizabeth Greig.
We also wish to thank the following: Maureen Laidlaw (tissue culture), Katherine Haslam and Medical Illustration, Addenbrooke's Hospital, Cambridge (photography), Chris Madren, Department of Genetics (sequencing), and our medical colleagues who have kindly referred patients to the vitreoretinal service at Addenbrooke's Hospital.