Article Text

Abstract
Objectives Silver–Russell syndrome (SRS) is an imprinted disorder characterised by intrauterine growth retardation, relative macrocephaly, failure to thrive, typical facial phenotype and frequent body asymmetry. Feeding difficulties are frequently noted, but no study described evolution of gastrointestinal signs during infancy and their management in SRS. The aim of this study was to describe these abnormalities in a large cohort of children with SRS.
Design We included 75 patients (median age 24.3 months (5.1–135.2)) in the study. We retrospectively analysed nutritional status before growth hormone therapy, the frequency of gastrointestinal signs, such as gastroesophageal reflux (GER), vomiting, constipation and feeding difficulties, and nutritional management.
Results Maternal uniparental disomy for chromosome 7 was found in 10 patients and 11p15 hypomethylation in 65 patients. Malnutrition (defined as a weight/expected weight for height ratio <80%) was detected in 70% of the children. Gastrointestinal signs were found in 77%, including severe vomiting before the age of 1 year in 50% of cases, persistent vomiting from the age of 1 year in 29% of cases and constipation in 20% of cases. Severe GER was diagnosed in 55% of children by 24 h oesophageal pH-metry. Feeding difficulties were described in 65% of cases, with indications for dietary enrichment in 49%. Enteral nutrition by gastrostomy was indicated in 22% of cases.
Conclusions Digestive signs (GER, constipation) and malnutrition are frequent in children with SRS. The systematic exploration and management of these signs are crucial to improve the nutritional status of these children before initiating growth hormone therapy.
- Silver Russell syndrome
- gastroesophageal reflux
- digestive manifestations
- malnutrition
- failure to thrive
Statistics from Altmetric.com
What is already known on this topic
-
Silver–Russell syndrome (SRS) is an imprinted disorder characterised by intrauterine growth retardation, relative macrocephaly, failure to thrive, a typical facial phenotype and body asymmetry.
-
Feeding difficulties are frequently noted in SRS.
-
Little is known about gastrointestinal signs and their management in SRS.
What this study adds
-
More than 70% of children with SRS display digestive signs or malnutrition.
-
Severe gastroesophageal reflux is the most frequent digestive sign, with persistent vomiting after the age of 1 year.
-
Systematic exploration and management of digestive signs and malnutrition are crucial before starting growth hormone therapy.
Introduction
Silver–Russell syndrome (SRS) was first described in 1953 in newborns with intrauterine and postnatal growth retardation associated with a triangular face and body asymmetry.1 Its incidence is evaluated to 1–30/100 000 cases per year. Two principal molecular mechanisms have been implicated in approximately 60% of cases2: maternal uniparental disomy of chromosome 7 (mUPD)3 in 5%–10% of cases and hypomethylation of imprinted control region 1 (ICR1) on chromosome 11p15 in 30%–63% of cases.4–7 The main clinical features of SRS are intrauterine growth retardation (IUGR), with a lack of catch-up growth after birth, and relative macrocephaly.6 ,8 Without growth hormone (GH) therapy, the spontaneous final height is about 151 cm in men and 140 cm in women.9 Other phenotypical abnormalities have been reported, such as facial abnormalities (prominent forehead and triangular face), body asymmetry, hypoglycaemia, café au lait spots or various skin pigmentary changes and malformations, including clinodactyly of the fifth finger (figure 1).10

Patients with Silver–Russell syndrome due to hypomethylation 11p15 (1A and 1B) and maternal uniparental disomy of chromosome 7 (2A and 2B, adapted from Wakeling EL13).
SRS, although fairly easy to recognise in extreme cases, may be difficult to diagnose in more subtle situations, particularly in the absence of body asymmetry. Many reports have highlighted certain similarities in the characteristics of patients with SRS. Not all authors agree on the cardinal signs of SRS for the establishment of a diagnosis.6 ,11 The main criteria used are being born small-for-gestational-age (SGA), with postnatal growth retardation, relative macrocephaly, body asymmetry and prominent forehead. Feeding difficulties are also included in a few clinical scores because they have been reported to be frequent during early childhood in patients with SRS.12 ,13 In a recent study of patients with genetically confirmed SRS, feeding difficulties were detected in 82% of these children versus only 48% of children who were SGA but did not have SRS (p=0.001).6 A body mass index Z-score <−2 SD score (SDS) was noted in 61% of children with SRS but in only 21% of children SGA without SRS (p<0.001), suggesting that malnutrition was also common.
In addition to feeding difficulties, gastrointestinal manifestations, such as constipation or severe vomiting, are frequently reported by the parents of children with SRS. Surprisingly, little is known about the severity and evolution during infancy of these signs. Only one study reported gastroesophageal reflux (GER) disease in 34% of these children and oesophagitis in 25%.14 In this study, SRS diagnosis was based purely on clinical features, with no genetic testing. As clinical diagnosis is difficult and not entirely reliable, due to phenotypical heterogeneity, it is crucial to confirm these findings in children with genetically confirmed SRS. The aim of this study was to describe the prevalence and management of gastrointestinal and nutritional signs in a cohort of 75 children with genetically confirmed SRS and to propose specific recommendations for their management.
Patients and methods
Patients
We retrospectively included 75 children with SRS born between 1982 and 2010. In all cases, the molecular diagnosis was made by the Molecular Biology Department of Trousseau Hospital. All patients or parents gave written informed consent for SRS analysis in accordance with French national ethics rules (Assistance Publique—Hôpitaux de Paris authorisation no. 682). Allele-specific methylated multiplex real-time quantitative PCR (ASMM RTQ-PCR) was used to analyse the methylation status of the 11p15 ICR1 domain and of the 11p15 ICR2 domain, as previously described.15 We assessed mUPD7 by analysing the methylation status of the imprinted PEG1/MEST region by ASMM RTQ-PCR as previously described.16 We collected clinical data on a medical questionnaire, which was completed by the referring clinician in each case. For children followed in our department, data were collected from medical records. The questionnaire included data about SRS phenotype, birth measurements, weight and length at the age of 2 years or as close to that age as possible independently of the age at diagnosis. We recorded the presence of digestive signs up to and after the age of 6 years. We classified GER in only the children with abnormal oesophageal pH-metry independently of clinical manifestations. The number of children receiving GH treatment and the age at which this treatment was initiated were also recorded.
Complementary methods and statistical analysis are described in online supplementary data.17–21
Results
The clinical characteristics of the 75 children with SRS are shown in table 1. As expected, most were SGA or displayed IUGR at birth, particularly in terms of their body length, with relative macrocephaly. Only two of the children with mUPD7 had a birth length and weight >−2 SDS. Nine children had symmetric IUGR, with head circumferences of <−2 SDS. Episodes of repeated symptomatic hypoglycaemia (defined by a blood glucose concentration below 60 mg/dL and confirmed by blood glucose tests during hospitalisation or after a malaise) occurred in nine children (12% of cases). Neurodevelopmental delay was noted in 15% of the children.
Molecular abnormalities and clinical characteristics of 75 patients with SRS
Postnatal growth retardation was evaluated at a median age of 2.7 years (0.4–11.2 years). All but one of the children had severe growth retardation with mean weight and/or length Z-scores <−2 SDS and 88% had a mean Z-score for weight below −3 SDS (table 2). By contrast to the measurements obtained at birth, weight Z-score was lower than length Z-score, suggesting a severe deficiency of weight gain during the first years of life. In addition, 46 children (65%) had malnutrition, with a W/H ratio <80%. As expected, feeding difficulties were frequently reported (67% of children with SRS). Nutritional support, such as an enriched diet, was indicated in 39% of children, and oral nutritional supplements were indicated in 33%. Enteral feeding was necessary in 30% of cases, with gastrostomy in 72% of them (n=16). Nissen fundoplication was associated to gastrostomy in only seven children due to severe GER.
Nutritional status of the 75 children with SRS according to epigenotype
The gastrointestinal signs reported are described in table 3. Clinical regurgitation or vomiting was reported in 50% of children before 1 year of age and persisted after 1 year of age in 29% of cases. Oesophageal pH monitoring over a period of 24 h was performed in only 24 children at a mean age of 3 years to check for GER, particularly in cases of frequent vomiting or malaise, or before surgery for gastrostomy. The results were considered pathological in more than half the children explored. Barium upper gastrointestinal X-ray was carried out in 21 children and revealed delayed gastric emptying in three cases and one case of intestinal malrotation. One child underwent surgery for intestinal volvulus on the common mesentery. Nissen fundoplication was performed in all children with severe GER resistant to medical treatment. Constipation was noted in 20% of patients, but only one child had severe signs requiring repeated cleansing enemas.
Gastrointestinal signs and their management in children with SRS by epigenotype and in healthy populations
No significant difference between molecular abnormalities was found for the phenotypes studied, other than weight Z-score, which was significantly lower in the children with 11p15 SRS (p=0.04). In children with development delay (15% of children), digestive manifestations were more frequent compared with the rest of the cohort. Indeed, in this subgroup, feeding difficulties and constipation were reported in 80% and 60% of cases, respectively. In addition, gastrostomy was more frequently indicated (60% of children) and associated to Nissen fundoplication in 40% of cases.
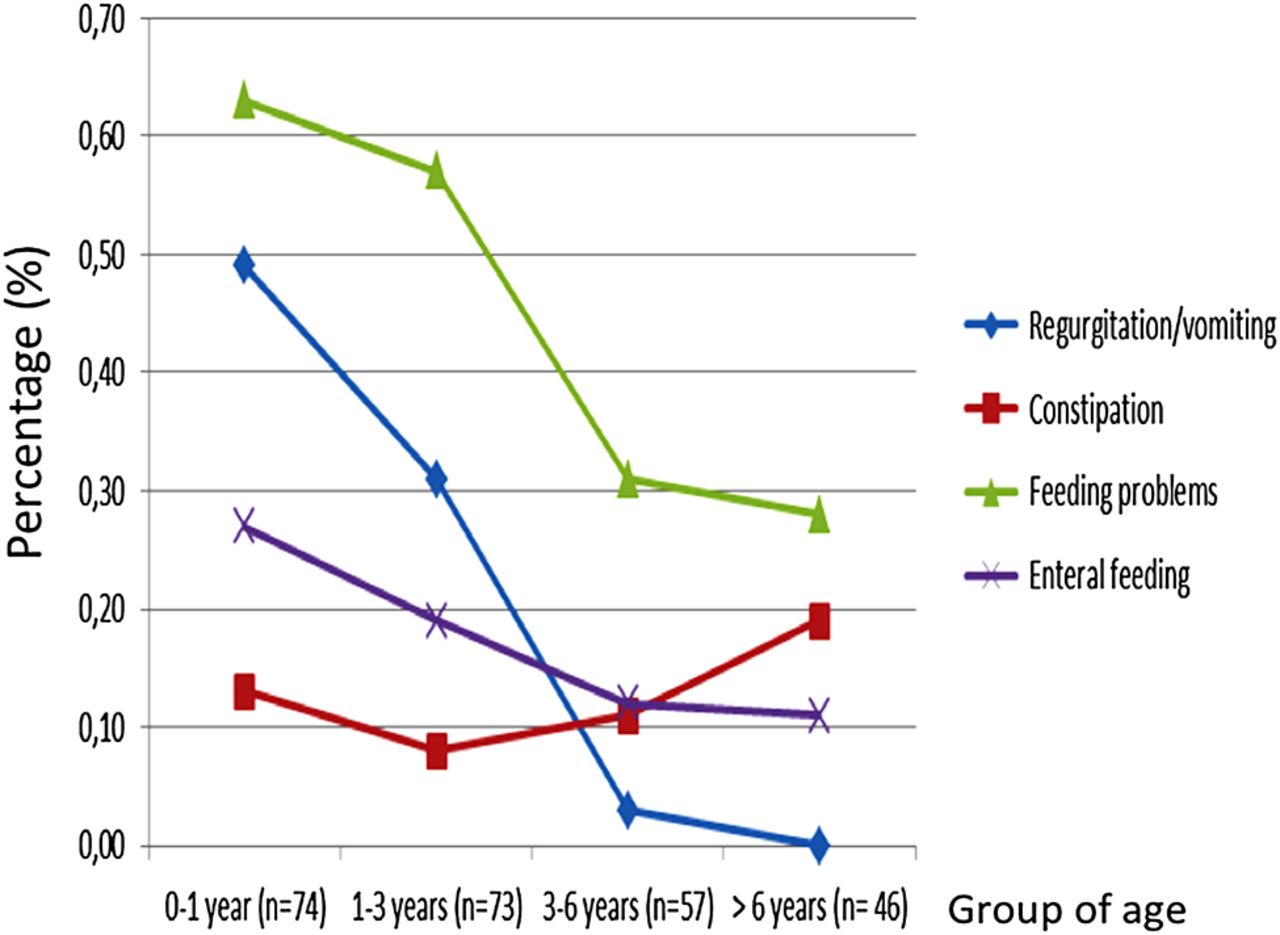
We evaluated changes in digestive signs from birth until the age of 6 years (figure 2). Feeding problems were more frequent in younger children, particularly before the age of 3 years (63% before the age of 1 year and 57% between the ages of 1 and 3 years). These problems tended to disappear after the age of 3 years. Regurgitations and vomiting also followed a similar course. By contrast, constipation tended to worsen after the age of 6 years (10% vs 19% of cases).
{kind=link}
{kind=link}
Evolution of gastrointestinal signs in children with Silver–Russell syndrome, from birth to the age of 6 years. The cohort was separated into four age groups (0–1 year/1–3years/3–6 years/over 6 years). We assessed the frequency of gastrointestinal signs by age group.
Discussion
The aim of this study was to assess digestive and nutritional signs and their current management retrospectively in a large cohort of 75 children with molecularly confirmed SRS. Such signs were found in 77% of these children, with regurgitation and/or vomiting before the age of 1 year reported most frequently. Malnutrition and feeding difficulties were also very common in these children.
In our cohort of children with SRS, regurgitations and/or vomiting, the most frequent digestive signs observed before the age of 1 year were twice as frequent as in the French general paediatric population at the same age.22 These signs may contribute to the observed postnatal growth retardation and feeding problems in some children.23 Oesophageal pH monitoring over 24 h yielded pathological results in >50% of the children explored, confirming the high frequency of GER at a mean age of 3 years. Surprisingly, two children with severe GER (reflux index >20%) displayed no clinical signs or atypical signs, such as recurrent abdominal pain, asthma or an unusual taste in the mouth (personal data), suggesting that the symptoms of GER may be atypical in children with SRS. Signs of GER tended to disappear in older children with SRS (>6 years of age) in our cohort, but severe undiagnosed GER might entail a risk of oesophageal complications (Barrett's oesophagitis, precancerous lesions) later in life.24 After exclusion of other diagnosis responsible for vomiting as cow's milk intolerance, we suggest that oesophageal pH monitoring over a period of 24 h (associated to oesophageal impedance study if possible) should be carried out in these children, particularly in cases of unusual GER manifestations (feeding problems, chronic abdominal pain or malnutrition), to ensure the early detection and treatment of GER.23 In addition, its control may contribute to improve the nutritional state of children with SRS.
Delayed gastric emptying or anatomic abnormalities, such as intestinal malrotation, may induce or aggravate GER.25 In our cohort, three of 21 barium upper gastrointestinal X-rays were abnormal. Further studies are required to determine whether such abnormalities are more frequent in children with SRS, but such abnormalities might result in a slow progression of food through the digestive tract, gastric dilatation after meals and an increase in the risk of vomiting or abdominal pain. They may also contribute to feeding difficulties and malnutrition, as described in other digestive diseases, such as chronic intestinal pseudo-obstruction.26 Finally, constipation is reported in 20% of older (>6 years) children in our cohort, a figure not significantly different from that reported for the general population.27 Overall, these findings suggest that children with SRS may present digestive motility disorder. Dysfunction of the upper oesophageal sphincter, with incomplete and/or uncoordinated upper oesophageal sphincter relaxation, has been described before.28 The tensin 3-gene, encoding a focal adhesion protein and located on chromosome 7, has recently been implicated in gut development in animal models, suggesting a potential role in SRS in patients with mUPD of chromosome 7.29
Feeding difficulties were identified in 67% of the patients in our cohort, confirming that these signs are potential major criteria for clinical diagnosis.6 Various mechanisms may be involved, such as oral motor dysfunction, which has been described in children with failure to thrive and growth disorders.30 The particular facial features (low cranial base height and mandibular body size associated with delayed dental age) observed in children with SRS may also play a role.31 ,32 Finally, dysregulation of the central control of food intake may be another causal factor.33 In humans, the IGF2 gene is located on chromosome 11p15.5, a region containing numerous imprinted genes. IGF2 is involved principally in regulating fetal growth and is expressed in several tissues, including the brain and the gut.34 The low level of expression of this gene in patients with SRS4 is responsible for the observed severe in utero growth retardation, but may also be involved in gut dysfunctions observed in SRS.
In our cohort, feeding difficulties tended to disappear during childhood, particularly after the age of 3 years. This observation may be explained by the positive effect of GH on food intake.35 However, as in Prader–Willi syndrome (PWS), changes in the secretion of appetite hormones during childhood may also be involved. Goldstone et al36 recently suggested that abnormal and/or delayed development or a sensitivity of the effector pathways of appetitive hormones, such as ghrelin, may interact with low levels of anorexigenic pancreatic polypeptides, contributing to the hyperphagia observed in late infancy in PWS. Further studies focusing on the hypothalamus–gut axis in SRS are now crucial to elucidate the mechanisms underlying these clinical manifestations.
Malnutrition affected 70% of the children with SRS in our cohort, particularly during early childhood. It probably results from the severe feeding difficulties and digestive signs. Pre-existing malnutrition may limit the beneficial effects of GH on height, so early nutritional management is probably crucial. Indeed, GH has metabolic effects, stimulating the secretion of IGF1, which is important for tissue anabolism.37 Nutritional status can be improved in several ways. Nutritional management, including the use of an enriched diet and/or enteral feeding, may also be considered. In our cohort, 30% of the children received enteral feeding, mostly for severe feeding difficulties with malnutrition, recurrent severe hypoglycaemia and/or major fatigability with an altered quality of life. Prospective studies are required to confirm the positive effect of nutritional management on the efficacy of GH for increasing the final adult height of patients with SRS. Indeed, metabolic disorders, probably due to excessive caloric intake, have been described later in life, in children with a history of IUGR.38 We generally recommend a W/H ratio target of about 80%–85%, but not higher, for the initiation of GH therapy, to be maintained throughout childhood and adolescence, to limit the risk of excess fat mass and metabolic abnormalities later in life. However, additional prospective studies are required to validate our current practice.
Gastrointestinal signs and nutritional state did not differ significantly between patients with different molecular abnormalities, except for weight Z-score, which was lower in children with 11p15 SRS. This suggests that an analysis of digestive signs cannot distinguish between children carrying the two known genetic abnormalities responsible for SRS (mUPD or hypomethylation of ICR1 on chromosome 11p15). More detailed phenotypical analysis is probably required, focusing on dysmorphology, the type of associated malformations and their frequency, together with cognitive development.
In conclusion, we found that gastrointestinal manifestations and malnutrition were frequent in children with genetically proved SRS. Even if our results need to be confirmed in prospective studies, we suggest that digestive explorations and nutritional evaluation should be systematically carried out as soon as possible after diagnosis to improve the management of these manifestations. Systematic and repeated evaluations of caloric intake are particularly crucial for consideration of the indications for nutritional support (enriched diet, oral nutritional supplements, enteral feeding) and to avoid malnutrition or overfeeding for these children. Indeed, children with SRS are characterised by abnormal body composition with low muscle mass (especially in young SRS) and variable amount of fat mass depending on nutritional status. Due to this particular situation, the cut-off for acceptable nutritional status is probably within a highly limited window (W/H ratio between 75% and 85%) depending on the importance of fat mass related to their low muscle mass. According to our experience, in case of SRS with W/H <75%, the extreme thinness limits probably growth velocity. But in case of overnutrition leading to ratio W/H >85%, children with SRS develop excessive fat mass related to their low muscle mass with higher risk of metabolic abnormalities, premature adrenarche, central puberty and short stature. Close collaboration between the paediatric endocrinologist and the gastroenterologist is particularly important for optimisation of the management of SRS, particularly before the initiation of GH therapy.
Acknowledgments
We thank Dr Emma Wakeling who provided the pictures of children with SRS in addition to the parents of the patients for agreeing to participate in this study (AFIF SSR/PAG Association FrançaIses des Familles ayant un enfant atteint du Syndrome Silver Russell (SSR) ou né Petit pour l'Age Gestationnel (PAG) et leurs amis) and the “Armand Trousseau Hospital SRS Research Group” including clinicians worldwide who kindly provided clinical details for their patients. List of the clinicians is available in supplementary data.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Collaborators V Abadie, S Alcayde, Y Alembik, J Amiel, G Baujat, C Baumann-Morel, E Bieth, AM Bertrand, D Bonneau, N Bouhours Nouet, C Brachet, F Brioude, JP Brossier, O Boute, S Cabrol, JC Carel, B Chabrol, O Chivu, P Christin, P Collignon, MP Cordier, V Cormier Daire, C Coubes, B Coupe, R Coutant, M Craen, H Crosnier, C De Baufort, A David, A Delahaye, B Delobel, MA Delrue, A Dieux Coeslier, MA Dommergues, B Doray, B Duban-Bedu, S Dufresne, P Edery, B Esteva, C Farges, I Fechtner, C Francannet, B Gilbert Dussardier, B Gilbert, E Ginglinger, F Giullano, A Goldenberg, O Hamiel, MD Harbison, C Heinrichs, D Heron, M Holder, M Houang, D Genevieve, M Gerard, M Gonzales, P Jantchou, P Jonveaux, PS Jouk, F Kurtz, Y Le Bouc, M Le Merrer, A Linglart, B Leheup, M Lebrun, J Leger, A Leinhart, GA Loeuille, S Manouvrier, D Martin- Coignard, JC Mas, M Mathieu, S Mercier, B Mignot, F Morice-Picard, G Morin, R Newfield, S Odent, I Oliver-Petit, L Olivier-Faivre, E Petriczko, N Philip, C Pienkowski, L Pinson, G Pinto, M Polak, C Quelin, M Port-lis, JC Reiter, M Rio, MF Riviere, B Roquelaure, J Salem, D Simon, S Soskin, Y Sznajer, M Tauber, C Thauvin, R Touraine, C Teinturier, A Toutain, L Van Maldergem, A Verloes, C Vincent Delorme, T-A Vu-Hong, J Weill.
-
Contributors CM collected the data, carried out the initial analyses, drafted the initial manuscript and approved the final manuscript as submitted. SR designed the data collection instruments, supervised data collection and approved the final manuscript as submitted. PT reviewed and revised the manuscript, and approved the final manuscript as submitted. IN designed the data collection instruments, reviewed and revised the manuscript, performed the molecular investigations, and approved the final manuscript as submitted. BD conceptualised and designed the study, designed data collection, carried out the initial analyses, reviewed and revised the manuscript, and approved the final manuscript as submitted.
-
Funding This work was supported by APHP, INSERM, UPMC-Paris funding, ANR EPIFEGRO. 2010.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval APHP.
-
Provenance and peer review Not commissioned; externally peer reviewed.