Article Text

Abstract
An Asian girl presented with failure to thrive, congenital hepatic fibrosis, protein losing enteropathy, and hypoglycaemia. Phosphomannose isomerase activity in skin fibroblasts was reduced. She is homozygous for a mutation, D131N, in the phosphomannose isomerase gene (PM1), consistent with the diagnosis of carbohydrate deficient glycoprotein syndrome type 1b. She responded to oral mannose treatment.
- carbohydrate deficient glycoprotein syndrome
- enteropathy
- hepatic fibrosis
- hyperinsulinism
- hypoglycaemia
- mannose
Statistics from Altmetric.com
- carbohydrate deficient glycoprotein syndrome
- enteropathy
- hepatic fibrosis
- hyperinsulinism
- hypoglycaemia
- mannose
The carbohydrate deficient glycoprotein syndromes (CDGS) are a group of disorders caused by genetically determined abnormalities of protein glycosylation.1 Glycosylation of proteins is fundamental to the functioning of many cellular processes, hence these conditions present with multisystemic symptoms. Global developmental delay is a major feature of all types of CDGS except for an emerging subgroup of children with a deficiency of phosphomannose isomerase (PMI), CDGS type 1b.2 These children have minimal neurological symptoms and suffer predominantly from gastrointestinal and hepatic disorders which appear to be reversible by supplementation of the diet with mannose.3 We describe the first recognised case of CDGS type 1b in the UK to highlight the importance of recognising this potentially lethal but treatable metabolic disorder.
Case report
An Asian girl of non-consanguineous parents presented at the age of 6 months with poor feeding, failure to thrive (weight 0.4th centile and height 2nd centile on 1996/1 United Kingdom growth chart) and mild developmental delay. Examination and routine investigations failed to find a specific cause. By 9 months of age her liver was enlarged and alanine aminotransferase was transiently increased to 887 IU/l (normal range less than 40 IU/l). The causes of chronic hepatitis were excluded and a liver biopsy showed the typical features of congenital hepatic fibrosis with very notable fibrous expansion of portal areas and mildly dilated ductal structures. Her next admissions were precipitated by increasingly severe episodes of diarrhoea and vomiting. Serum albumin was low at 18 g/l (normal range 36–51 g/l) and did not recover between episodes; her stools remained loose. She had no proteinuria. A small bowel biopsy showed partial villous atrophy and some crypt hyperplasia, but no dilated lacteals; she had no serological evidence of coeliac disease. Hypoglycaemia was noted during these episodes of diarrhoea and vomiting and was associated with hyperinsulinism.
During further investigation for metabolic disease, analysis of serum transferrin by agarose gel electrophoresis revealed the classical pattern of CDGS type 1 (fig 1). Clearly she did not have the neurological features of CDGS type 1a, and a magnetic resonance imaging scan of her brain was normal. Secondary causes of abnormal glycosylation caused by galactosaemia and hereditary fructose intolerance were excluded. In retrospect she did have the characteristic fat pads on her chin and thighs as well as the inverted nipples described with CDGS type 1a. A thrombotic tendency screen was abnormal with decreased concentrations of antithrombin III and protein C.

Effects of oral mannose on transferrin glycoforms of patient with CDGS type 1b. Lanes: (1) normal serum; (2) normal cerebrospinal fluid; (3) serum of father who is a carrier; (4) serum of mother, also a carrier; (5) (6/98) pretreatment sample from patient; (6)–(8) post-treatment samples showing fading of the bands representing asialotransferrin (arrow) and disialotransferrin (middle band) and increasing density of the tetrasialotransferrin (top band) towards normal. Post-neuraminidase samples in the same sequence prove that the charge heterogeneity cannot be caused by difference in the primary sequence of amino acids of the protein—that is, a sequence polymorphism.
The diagnosis of CDGS type 1b was made by enzyme analysis of her skin fibroblasts which showed normal activity of phosphomannomutase but only 30% of the phosphomannose isomerase activity of normal control fibroblasts. This diagnosis was confirmed by showing that she was homozygous for the mutation D131N in the PM1gene which encodes phosphomannose isomerase. Her parents are heterozygous for the mutation.
Treatment with oral mannose (SHS International Ltd, Liverpool, UK) was started at 100 mg/kg/dose five times daily, increased after seven days to 150 mg/kg/dose five times daily with no ill effect. On treatment her episodes of diarrhoea and vomiting became less severe and protracted. She did not become hypoglycaemic and could be managed at home with oral electrolyte solutions and mannose. Her thrombotic tendency screen returned to normal within 10 days of commencing mannose and over the next five months her transferrin pattern also improved (fig 1). Her growth (weight 25th centile and height between the 9th and 25th centiles on 1996/1 United Kingdom growth chart) and development are progressing normally now at age 4 years. Poor oral intake of food persisted for the first 15 months of treatment but has now normalised. She still has notable hepatomegaly (9 cm in epigastrium and 4 cm in the midclavicular line) but no splenomegaly or features of portal hypertension.
Discussion
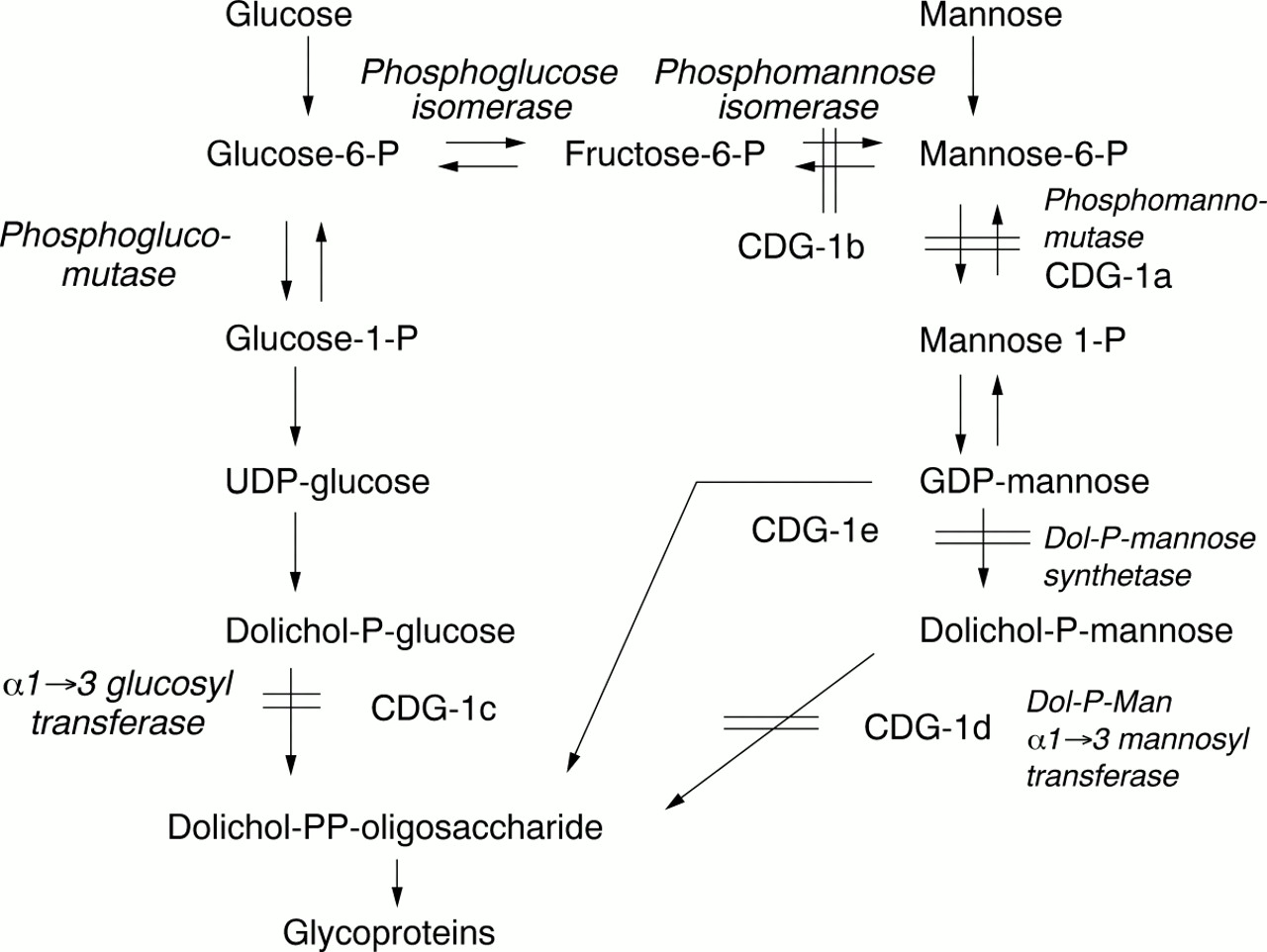
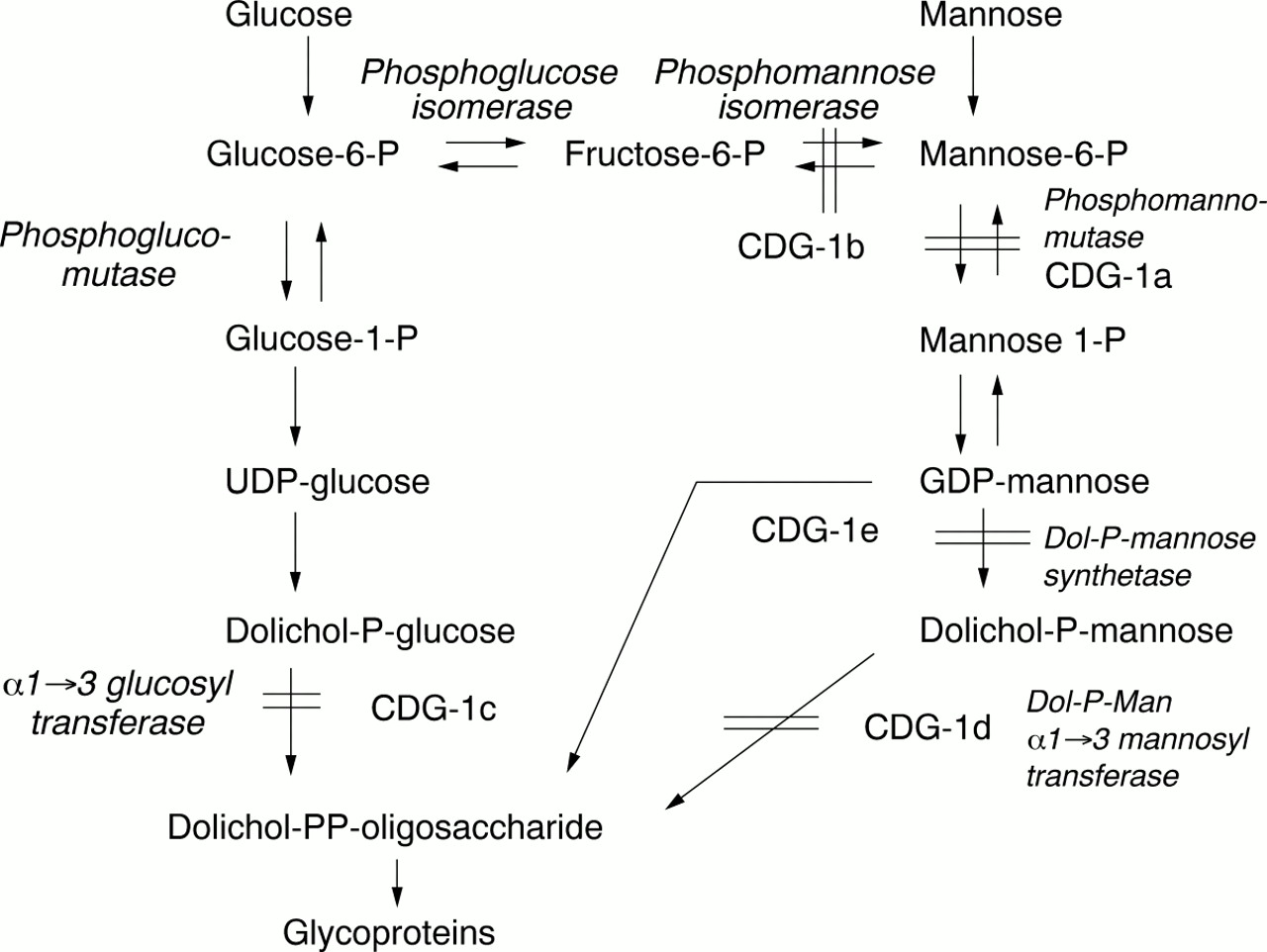
CDGS was first described by Jaeken et al in 1980, as a multisystem familial disorder associated with psychomotor retardation and an abnormal pattern of glycoproteins as detected on agarose gel electrophoresis.4 Transferrin is the most commonly used glycoprotein for biochemical testing. Initially four subgroups of CDGS were classified according to the pattern of serum transferrin glycoforms and clinical presentation.1The majority of cases were CDGS type 1 and most had a deficiency of phosphomannomutase (PMM) and mutations in thePMM2 gene. Clinical features included profound psychomotor retardation, hypotonia, failure to thrive, inverted nipples, unusual fat deposits, abnormal liver function tests, hepatomegaly, coagulopathy, and stroke like episodes. Neuroradiological imaging showed olivopontocerebellar degeneration. In 1998, when we were investigating our patient, Niehues et aldescribed a new type of CDGS with no neurological symptoms but with the same pattern of hypoglycosolation of transferrin as CDGS type 1.3 The patient had normal amounts of PMM but was deficient in phosphomannose isomerase (PMI), the enzymatic step before PMM (fig 2). This deficiency of PMI was classified as CDGS type 1b, and PMM deficiency as CDGS type 1a.
{kind=link}
{kind=link}
Metabolic pathway showing all the carbohydrate deficient glycoprotein syndromes. Note the important position of the PMI and PMM enzymes and the entry of mannose into the pathway to suggest why treatment should be effective.
At the time of writing, we are aware of nine cases of CDGS type 1b, with PMI deficiency, described in the literature. The clinical features have been tabulated in a recent paper and include ductal plate abnormalities of the liver (congenital hepatic fibrosis, Caroli syndrome), recurrent episodes of vomiting and diarrhoea, protein losing enteropathy, low serum albumin, failure to thrive, and hyperinsulinaemic hypoglycaemia.2 Most patients have had abnormalities of coagulation resulting in bleeding or a thrombotic tendency, and some have suffered serious thrombotic events. Only one other case of PMI deficiency had inverted nipples and our patient is the only case in which fat pads of the chin and thighs are described. These features are more typical of CDGS type 1a.
In 1996, Panneerselvam and Freeze5 reported that exogenous mannose transiently corrected altered N-glycosylation in fibroblasts from patients with CDGS type 1. Alton and colleagues6showed the feasibility and safety of orally ingested mannose as a treatment for CDGS and in 1998, Niehues and colleagues3successfully treated a patient with CDGS type 1b by using oral mannose. As in our patient, the clinical response to mannose was within two weeks; however, the improvement in glycosylation of glycoproteins was slower than in our experience. It is too early to predict the long term effects of this treatment, especially with respect to progression of the liver disease.
We publish this case report to highlight the importance of screening for CDGS in patients with hepatomegaly, hypoglycaemia, and gastrointestinal symptoms without neurological signs. Phosphomannose isomerase deficiency is probably underdiagnosed. The test is inexpensive and dietary treatment successful.
Acknowledgments
We would like to acknowledge Dr S Green for referring the patient and continuing her shared care. F Imtiaz acknowledges the receipt of a research studentship from the Saudi Arabian Cultural Bureau, London.